Arnadottir Gudny A, Jensson Brynjar O, Marelsson Sigurdur E, Sulem Gerald, Oddsson Asmundur, Kristjansson Ragnar P, Benonisdottir Stefania, Gudjonsson Sigurjon A, Masson Gisli, Thorisson Gudmundur A, Saemundsdottir Jona, Magnusson Olafur Th, Jonasdottir Adalbjorg, Jonasdottir Aslaug, Sigurdsson Asgeir, Gudbjartsson Daniel F, Thorsteinsdottir Unnur, Arngrimsson Reynir, Sulem Patrick, Stefansson Kari
deCODE Genetics/Amgen, Inc., Sturlugata 8, 101, Reykjavik, Iceland.
Department of Pediatrics, Landspitali University Hospital, Reykjavik, Iceland.
BMC Med Genet. 2017 Oct 2;18(1):103. doi: 10.1186/s12881-017-0466-8.
Epileptic encephalopathies are a group of childhood epilepsies that display high phenotypic and genetic heterogeneity. The recent, extensive use of next-generation sequencing has identified a large number of genes in epileptic encephalopathies, including UBA5 in which biallelic mutations were first described as pathogenic in 2016 (Colin E et al., Am J Hum Genet 99(3):695-703, 2016. Muona M et al., Am J Hum Genet 99(3):683-694, 2016). UBA5 encodes an activating enzyme for a post-translational modification mechanism known as ufmylation, and is the first gene from the ufmylation pathway that is linked to disease.
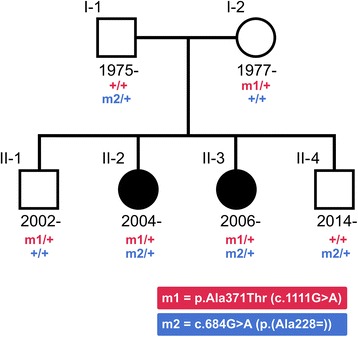
We sequenced the genomes of two sisters with early-onset epileptic encephalopathy along with their unaffected parents in an attempt to find a genetic cause for their condition. The sisters, born in 2004 and 2006, presented with infantile spasms at six months of age, which later progressed to recurrent, treatment-resistant seizures. We detected a compound heterozygous genotype in UBA5 in the sisters, a genotype not seen elsewhere in an Icelandic reference set of 30,067 individuals nor in public databases. One of the mutations, c.684G > A, is a paternally inherited exonic splicing mutation, occuring at the last nucleotide of exon 7 of UBA5. The mutation is predicted to disrupt the splice site, resulting in loss-of-function of one allele of UBA5. The second mutation is a maternally inherited missense mutation, p.Ala371Thr, previously reported as pathogenic when in compound heterozygosity with a loss-of-function mutation in UBA5 and is believed to produce a hypomorphic allele. Supportive of this, we have identified three adult Icelanders homozygous for the p.Ala371Thr mutation who show no signs of neurological disease.
We describe compound heterozygous mutations in the UBA5 gene in two sisters with early-onset epileptic encephalopathy. To our knowledge, this is the first description of mutations in UBA5 since the initial discovery that pathogenic biallelic variants in the gene cause early-onset epileptic encephalopathy. We further provide confirmatory evidence that p.Ala371Thr is a hypomorphic mutation, by presenting three adult homozygotes who show no signs of neurological disease.
癫痫性脑病是一组儿童癫痫疾病,具有高度的表型和遗传异质性。近年来,下一代测序技术的广泛应用已在癫痫性脑病中鉴定出大量基因,其中包括泛素样修饰激活酶5(UBA5)基因,2016年首次报道该基因的双等位基因突变具有致病性(科林·E等人,《美国人类遗传学杂志》99(3):695 - 703,2016年。莫纳·M等人,《美国人类遗传学杂志》99(3):683 - 694,2016年)。UBA5编码一种参与称为泛素样修饰(ufmylation)的翻译后修饰机制的激活酶,是泛素样修饰途径中首个与疾病相关的基因。
我们对两名患有早发性癫痫性脑病的姐妹及其未患病的父母进行了基因组测序,试图找出她们病情的遗传原因。这两名姐妹分别出生于2004年和2006年,6个月大时出现婴儿痉挛症,随后发展为反复发作且难以治疗的癫痫。我们在这两名姐妹中检测到UBA5基因的复合杂合基因型,在冰岛30,067人的参考样本中以及公共数据库中均未发现这种基因型。其中一个突变,c.684G > A,是父系遗传的外显子剪接突变,发生在UBA5基因第7外显子的最后一个核苷酸处。该突变预计会破坏剪接位点,导致UBA5的一个等位基因功能丧失。第二个突变是母系遗传的错义突变,p.Ala371Thr,先前报道当与UBA5的功能丧失突变呈复合杂合状态时具有致病性,据信会产生一个低表达等位基因。支持这一观点的是,我们发现了三名p.Ala371Thr突变纯合的成年冰岛人,他们没有神经系统疾病的迹象。
我们描述了两名患有早发性癫痫性脑病的姐妹中UBA5基因的复合杂合突变。据我们所知,这是自最初发现该基因的致病性双等位基因变异导致早发性癫痫性脑病以来,对UBA5基因突变的首次描述。我们还通过展示三名没有神经系统疾病迹象的成年纯合子,进一步提供了确证性证据,证明p.Ala371Thr是一种低表达突变。