Department of Computing, Imperial College London, South Kensington Campus, London, UK.
Cardiovascular Magnetic Resonance Imaging and Genetics, MRC London Institute of Medical Sciences, Imperial College London, Hammersmith Hospital Campus, London, UK.
Bioinformatics. 2018 Jan 1;34(1):97-103. doi: 10.1093/bioinformatics/btx552.
Left ventricular (LV) hypertrophy is a strong predictor of cardiovascular outcomes, but its genetic regulation remains largely unexplained. Conventional phenotyping relies on manual calculation of LV mass and wall thickness, but advanced cardiac image analysis presents an opportunity for high-throughput mapping of genotype-phenotype associations in three dimensions (3D).
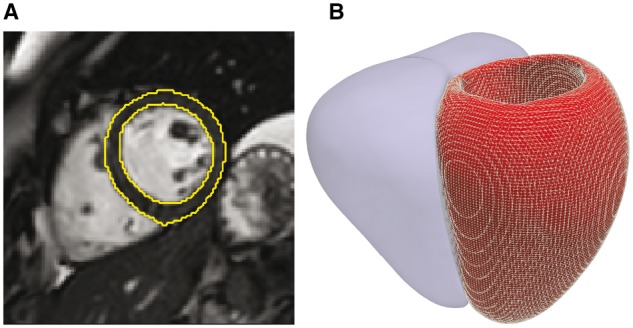
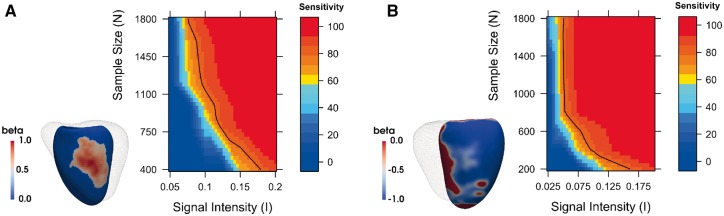
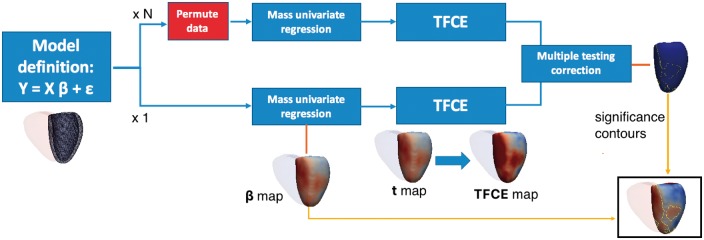
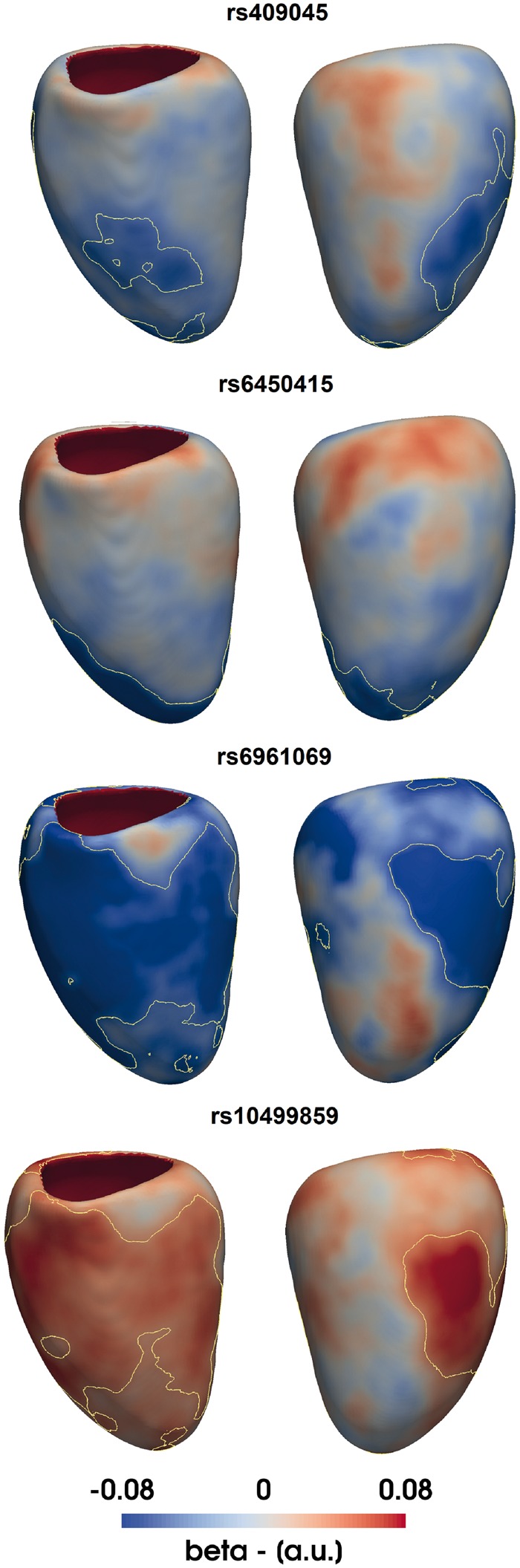
High-resolution cardiac magnetic resonance images were automatically segmented in 1124 healthy volunteers to create a 3D shape model of the heart. Mass univariate regression was used to plot a 3D effect-size map for the association between wall thickness and a set of predictors at each vertex in the mesh. The vertices where a significant effect exists were determined by applying threshold-free cluster enhancement to boost areas of signal with spatial contiguity. Experiments on simulated phenotypic signals and SNP replication show that this approach offers a substantial gain in statistical power for cardiac genotype-phenotype associations while providing good control of the false discovery rate. This framework models the effects of genetic variation throughout the heart and can be automatically applied to large population cohorts.
The proposed approach has been coded in an R package freely available at https://doi.org/10.5281/zenodo.834610 together with the clinical data used in this work.
Supplementary data are available at Bioinformatics online.
左心室(LV)肥大是心血管结果的强有力预测因子,但它的遗传调控在很大程度上仍未得到解释。传统表型依赖于 LV 质量和壁厚度的手动计算,但先进的心脏图像分析为在三维(3D)中进行基因型-表型关联的高通量映射提供了机会。
在 1124 名健康志愿者中,自动对高分辨率心脏磁共振图像进行分割,以创建心脏的 3D 形状模型。使用质量单变量回归在网格的每个顶点处绘制壁厚度与一组预测因子之间的关联的 3D 效应大小图。通过应用无阈值聚类增强来提高具有空间连续性的信号区域,确定存在显著效应的顶点。对模拟表型信号和 SNP 复制的实验表明,这种方法在提供良好的假发现率控制的同时,为心脏基因型-表型关联提供了统计功效的显著提高。该框架在整个心脏中模拟遗传变异的影响,并且可以自动应用于大型人群队列。
所提出的方法已在 R 包中进行编码,并可在 https://doi.org/10.5281/zenodo.834610 上免费获得,以及本工作中使用的临床数据。
补充数据可在“Bioinformatics”在线获得。