Yang Qian, Sing-Long Carlos A, Reed Evan J
Institute for Computational and Mathematical Engineering , Stanford University , Stanford , 94305 , USA . Email:
Mathematical and Computational Engineering , School of Engineering , Pontificia Universidad Catolica de Chile , Santiago , Chile . Email:
Chem Sci. 2017 Aug 1;8(8):5781-5796. doi: 10.1039/c7sc01052d. Epub 2017 Jun 19.
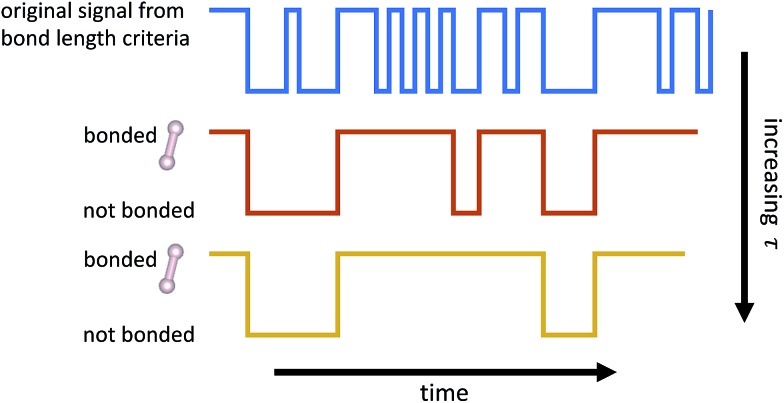
We propose a novel statistical learning framework for automatically and efficiently building reduced kinetic Monte Carlo (KMC) models of large-scale elementary reaction networks from data generated by a single or few molecular dynamics simulations (MD). Existing approaches for identifying species and reactions from molecular dynamics typically use bond length and duration criteria, where bond duration is a fixed parameter motivated by an understanding of bond vibrational frequencies. In contrast, we show that for highly reactive systems, bond duration should be a model parameter that is chosen to maximize the predictive power of the resulting statistical model. We demonstrate our method on a high temperature, high pressure system of reacting liquid methane, and show that the learned KMC model is able to extrapolate more than an order of magnitude in time for key molecules. Additionally, our KMC model of elementary reactions enables us to isolate the most important set of reactions governing the behavior of key molecules found in the MD simulation. We develop a new data-driven algorithm to reduce the chemical reaction network which can be solved either as an integer program or efficiently using L1 regularization, and compare our results with simple count-based reduction. For our liquid methane system, we discover that rare reactions do not play a significant role in the system, and find that less than 7% of the approximately 2000 reactions observed from molecular dynamics are necessary to reproduce the molecular concentration over time of methane. The framework described in this work paves the way towards a genomic approach to studying complex chemical systems, where expensive MD simulation data can be reused to contribute to an increasingly large and accurate genome of elementary reactions and rates.
我们提出了一种新颖的统计学习框架,用于从单个或少数分子动力学模拟(MD)生成的数据中自动高效地构建大规模基元反应网络的简化动力学蒙特卡罗(KMC)模型。现有的从分子动力学中识别物种和反应的方法通常使用键长和持续时间标准,其中键持续时间是基于对键振动频率的理解而设定的固定参数。相比之下,我们表明,对于高反应性系统,键持续时间应是一个模型参数,其选择应使所得统计模型的预测能力最大化。我们在高温高压下反应的液态甲烷系统上展示了我们的方法,并表明所学习的KMC模型能够对关键分子的时间进行超过一个数量级的外推。此外,我们的基元反应KMC模型使我们能够分离出在MD模拟中控制关键分子行为的最重要反应集。我们开发了一种新的数据驱动算法来简化化学反应网络,该网络既可以作为整数规划求解,也可以使用L1正则化有效地求解,并将我们的结果与基于简单计数的简化方法进行比较。对于我们的液态甲烷系统,我们发现稀有反应在该系统中不起重要作用,并且发现从分子动力学中观察到的大约2000个反应中,不到7%是重现甲烷随时间的分子浓度所必需的。这项工作中描述的框架为研究复杂化学系统的基因组方法铺平了道路,在这种方法中,昂贵的MD模拟数据可以被重新利用,以构建一个越来越大且准确的基元反应和速率基因组。