Zielinski François, Maxwell Peter I, Fletcher Timothy L, Davie Stuart J, Di Pasquale Nicodemo, Cardamone Salvatore, Mills Matthew J L, Popelier Paul L A
Manchester Institute of Biotechnology (MIB), 131 Princess Street, Manchester, M1 7DN, United Kingdom.
School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, United Kingdom.
Sci Rep. 2017 Oct 9;7(1):12817. doi: 10.1038/s41598-017-12600-3.
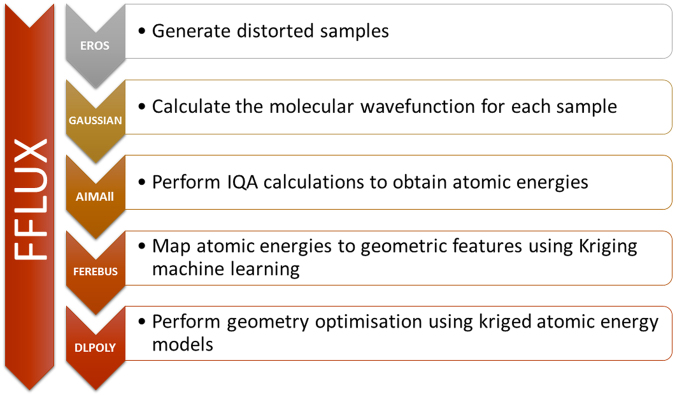
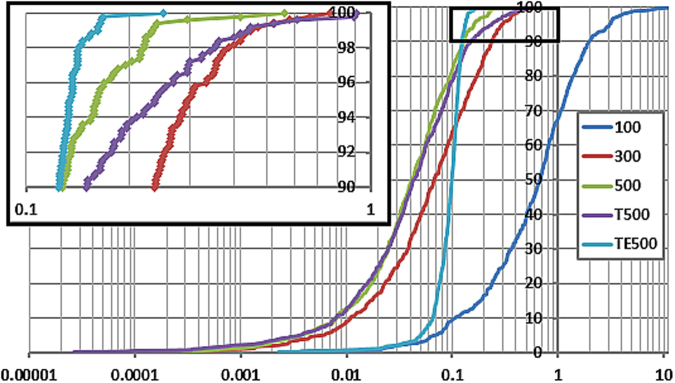
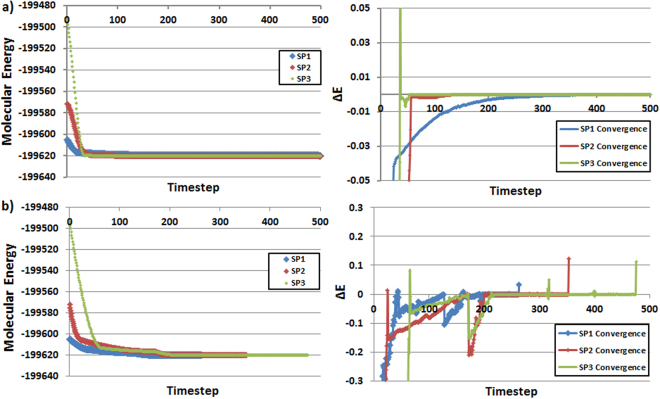
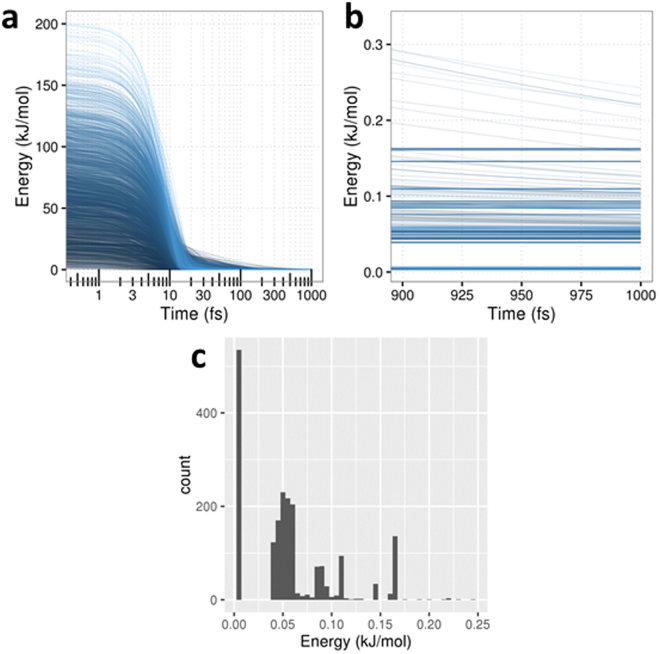
The geometry optimization of a water molecule with a novel type of energy function called FFLUX is presented, which bypasses the traditional bonded potentials. Instead, topologically-partitioned atomic energies are trained by the machine learning method kriging to predict their IQA atomic energies for a previously unseen molecular geometry. Proof-of-concept that FFLUX's architecture is suitable for geometry optimization is rigorously demonstrated. It is found that accurate kriging models can optimize 2000 distorted geometries to within 0.28 kJ mol of the corresponding ab initio energy, and 50% of those to within 0.05 kJ mol. Kriging models are robust enough to optimize the molecular geometry to sub-noise accuracy, when two thirds of the geometric inputs are outside the training range of that model. Finally, the individual components of the potential energy are analyzed, and chemical intuition is reflected in the independent behavior of the three energy terms Formula: see text, [Formula: see text] (electrostatic) and [Formula: see text] (exchange), in contrast to standard force fields.
本文介绍了一种利用名为FFLUX的新型能量函数对水分子进行几何优化的方法,该方法绕过了传统的键合势。相反,通过机器学习方法克里金法训练拓扑划分的原子能量,以预测其在先前未见分子几何结构下的IQA原子能量。严格证明了FFLUX架构适用于几何优化的概念验证。研究发现,精确的克里金模型可以将2000个扭曲的几何结构优化到与相应的从头算能量相差0.28 kJ/mol以内,其中50%优化到0.05 kJ/mol以内。当三分之二的几何输入超出该模型的训练范围时,克里金模型足够稳健,能够将分子几何结构优化到亚噪声精度。最后,分析了势能的各个组成部分,与标准力场相比,化学直觉体现在三个能量项(原子内)、(静电)和(交换)的独立行为中。