Institute for Advanced Biosciences, Keio University, Mizukami 246-2, Kakuganji, Tsuruoka, Yamagata, 997-0052, Japan.
BMC Genomics. 2017 Oct 13;18(1):784. doi: 10.1186/s12864-017-4162-z.
The reduced cost of sequencing has made de novo sequencing and the assembly of draft microbial genomes feasible in any ordinary biology lab. However, the process of finishing and completing the genome remains labor-intensive and computationally challenging in some cases, such as in the study of complete genome sequences, genomic rearrangements, long-range syntenic relationships, and structural variations.
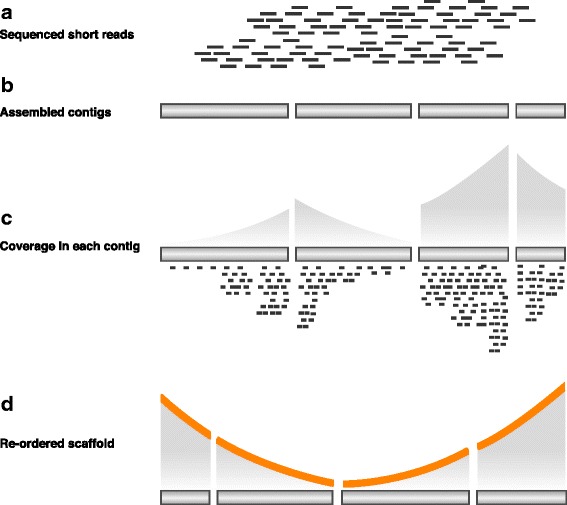
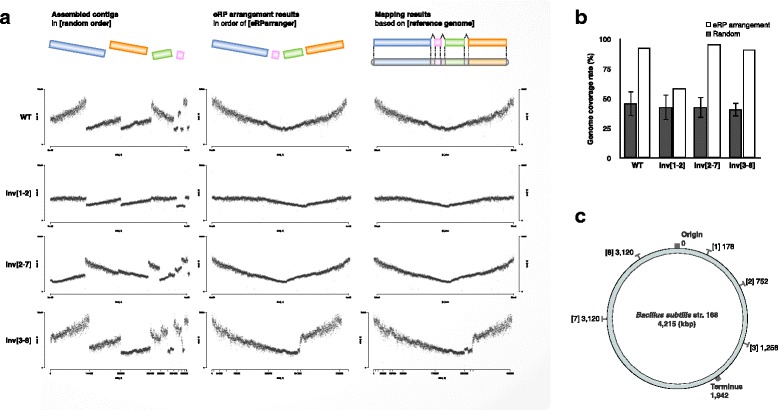
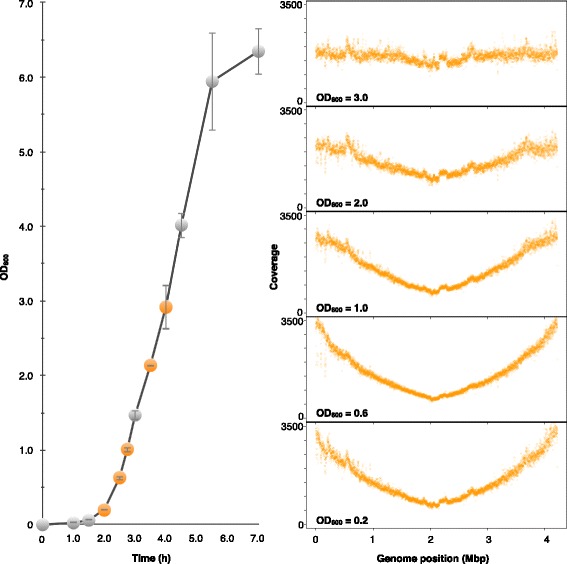
Here, we show a contig reordering strategy based on experimental replication profiling (eRP) to recapitulate the bacterial genome structure within draft genomes. During the exponential growth phase, the majority of bacteria show a global genomic copy number gradient that is enriched near the replication origin and gradually declines toward the terminus. Therefore, if genome sequencing is performed with appropriate timing, the short-read coverage reflects this copy number gradient, providing information about the contig positions relative to the replication origin and terminus.
We therefore investigated the appropriate timing for genomic DNA sampling and developed an algorithm for the reordering of the contigs based on eRP. As a result, this strategy successfully recapitulates the genomic structure of various structural mutants with draft genome sequencing.
Our strategy was successful for contig rearrangement with intracellular DNA replication behavior mechanisms and can be applied to almost all bacteria because the DNA replication system is highly conserved. Therefore, eRP makes it possible to understand genomic structural information and long-range syntenic relationships using a draft genome that is based on short reads.
测序成本的降低使得从头测序和 draft 微生物基因组的组装在任何普通的生物学实验室中都成为可能。然而,在某些情况下,如对完整基因组序列、基因组重排、长程同线性关系和结构变异的研究,完成和完成基因组的过程仍然是劳动密集型和计算挑战性的。
在这里,我们展示了一种基于实验复制分析 (eRP) 的 contig 重排策略,以重现 draft 基因组中的细菌基因组结构。在指数生长阶段,大多数细菌表现出一个全局基因组拷贝数梯度,该梯度在复制原点附近富集,并逐渐向末端下降。因此,如果在适当的时间进行基因组测序,短读测序的覆盖度反映了这个拷贝数梯度,提供了关于 contig 相对于复制原点和末端位置的信息。
因此,我们研究了基因组 DNA 采样的适当时间,并基于 eRP 开发了一个 contig 重排的算法。结果,该策略成功地重现了具有 draft 基因组测序的各种结构突变体的基因组结构。
我们的策略对于基于细胞内 DNA 复制行为机制的 contig 重排是成功的,并且几乎可以应用于所有细菌,因为 DNA 复制系统高度保守。因此,eRP 使得使用基于短读的 draft 基因组来理解基因组结构信息和长程同线性关系成为可能。