Kamath Govinda M, Shomorony Ilan, Xia Fei, Courtade Thomas A, Tse David N
Department of Electrical Engineering, Stanford University, Stanford, California 94305, USA.
Department of Electrical Engineering and Computer Sciences, University of California, Berkeley, California 94720, USA.
Genome Res. 2017 May;27(5):747-756. doi: 10.1101/gr.216465.116. Epub 2017 Mar 20.
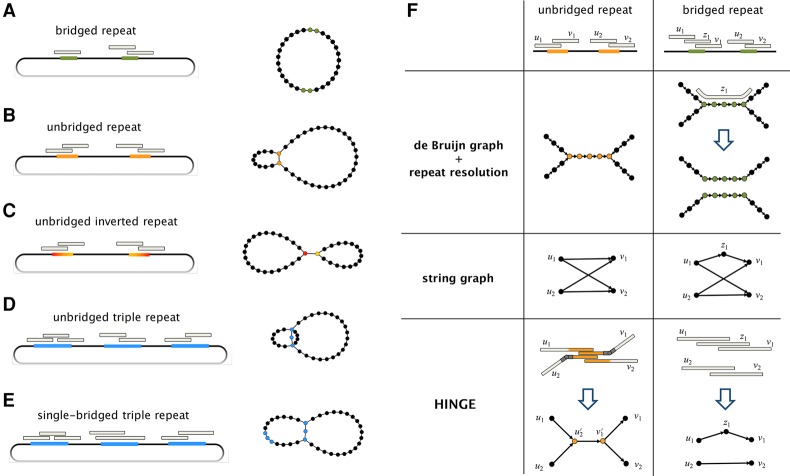
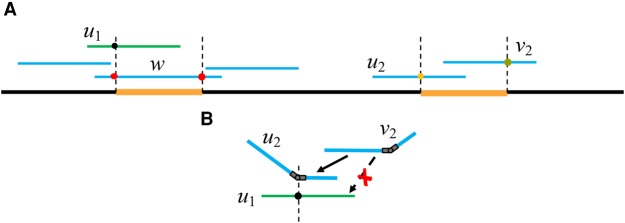
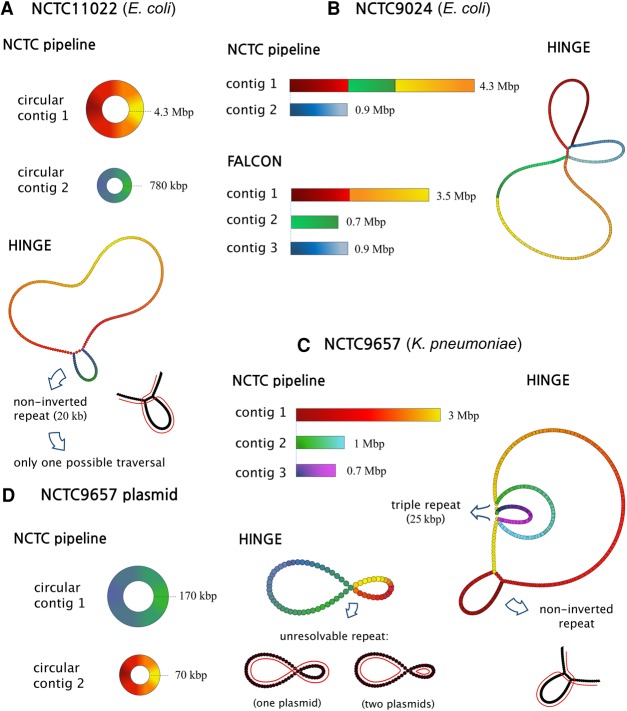
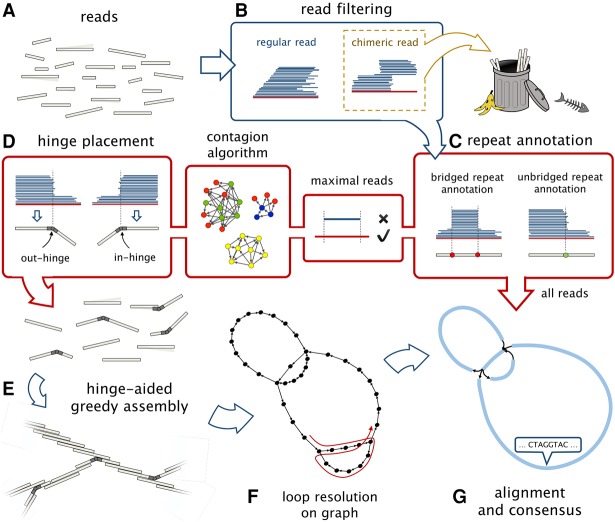
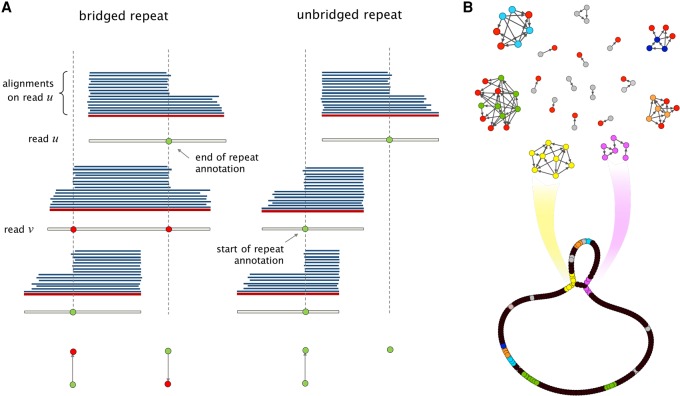
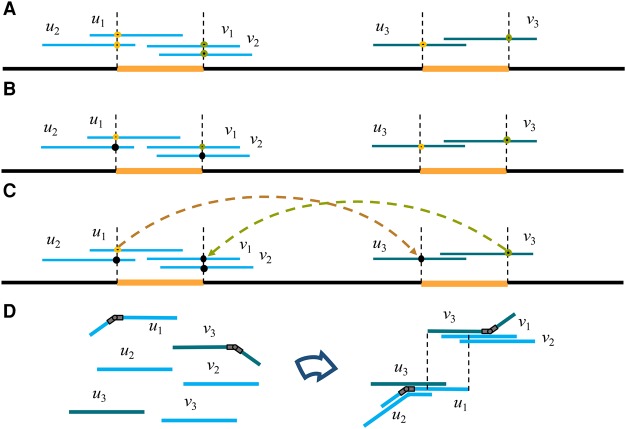
Long-read sequencing technologies have the potential to produce gold-standard de novo genome assemblies, but fully exploiting error-prone reads to resolve repeats remains a challenge. Aggressive approaches to repeat resolution often produce misassemblies, and conservative approaches lead to unnecessary fragmentation. We present HINGE, an assembler that seeks to achieve optimal repeat resolution by distinguishing repeats that can be resolved given the data from those that cannot. This is accomplished by adding "hinges" to reads for constructing an overlap graph where only unresolvable repeats are merged. As a result, HINGE combines the error resilience of overlap-based assemblers with repeat-resolution capabilities of de Bruijn graph assemblers. HINGE was evaluated on the long-read bacterial data sets from the NCTC project. HINGE produces more finished assemblies than Miniasm and the manual pipeline of NCTC based on the HGAP assembler and Circlator. HINGE also allows us to identify 40 data sets where unresolvable repeats prevent the reliable construction of a unique finished assembly. In these cases, HINGE outputs a visually interpretable assembly graph that encodes all possible finished assemblies consistent with the reads, while other approaches such as the NCTC pipeline and FALCON either fragment the assembly or resolve the ambiguity arbitrarily.
长读长测序技术有潜力生成金标准的从头基因组组装结果,但充分利用易出错的读段来解析重复序列仍是一项挑战。激进的重复序列解析方法往往会产生错误组装,而保守的方法则会导致不必要的片段化。我们提出了HINGE,这是一种组装程序,旨在通过区分根据数据可解析的重复序列和不可解析的重复序列来实现最佳的重复序列解析。这是通过在读取序列中添加“铰链”来构建重叠图来实现的,在该重叠图中,只有不可解析的重复序列才会被合并。因此,HINGE将基于重叠的组装程序的错误恢复能力与德布鲁因图组装程序的重复序列解析能力结合起来。HINGE在来自NCTC项目的长读长细菌数据集上进行了评估。与基于HGAP组装程序和Circlator的Miniasm以及NCTC的手动流程相比,HINGE产生的完整组装结果更多。HINGE还使我们能够识别40个数据集,在这些数据集中,不可解析的重复序列阻碍了唯一完整组装的可靠构建。在这些情况下,HINGE输出一个可视化可解释的组装图,该图编码了与读段一致的所有可能的完整组装结果,而其他方法,如NCTC流程和FALCON,要么使组装碎片化,要么任意解决模糊性问题。