Croxen Matthew A, Macdonald Kimberley A, Walker Matthew, deWith Nancy, Zabek Erin, Peterson Christy, Reimer Aleisha, Chui Linda, Tschetter Lorelee, Hoang Linda, King Robin K
British Columbia Centre for Disease Control Public Health Laboratory, Vancouver, British Columbia, Canada.
National Microbiology Laboratory (NML), Winnipeg, Manitoba, Canada; British Columbia Centre for Disease Control (BCCDC) Public Health Laboratory, Vancouver, British Columbia, Canada.
PLoS Curr. 2017 Aug 9;9:ecurrents.outbreaks.309af53b9edcc785163539c30c3953f6. doi: 10.1371/currents.outbreaks.309af53b9edcc785163539c30c3953f6.
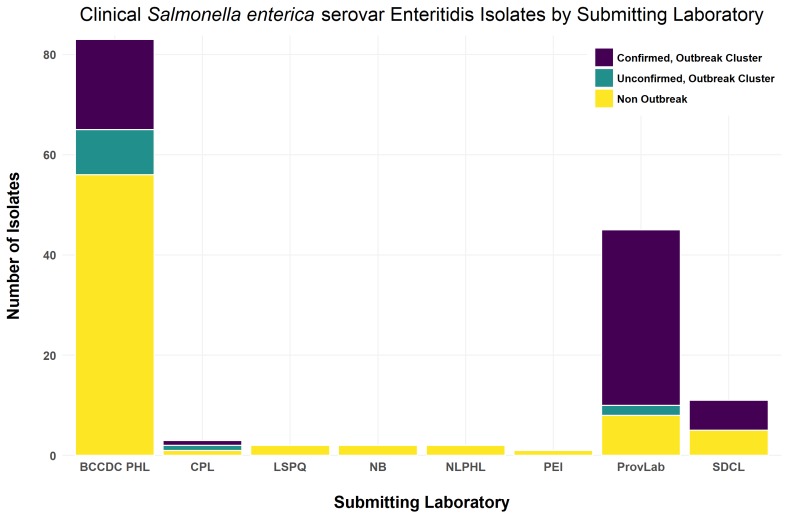
A multi-provincial outbreak of Salmonella enterica serovar Enteritidis was linked to newly hatched chicks and poults from a single hatchery during the spring of 2015. In total, there were 61 human cases that were epidemiologically confirmed to be linked to the chicks and poults and the outbreak was deemed to have ended in the summer of 2015.
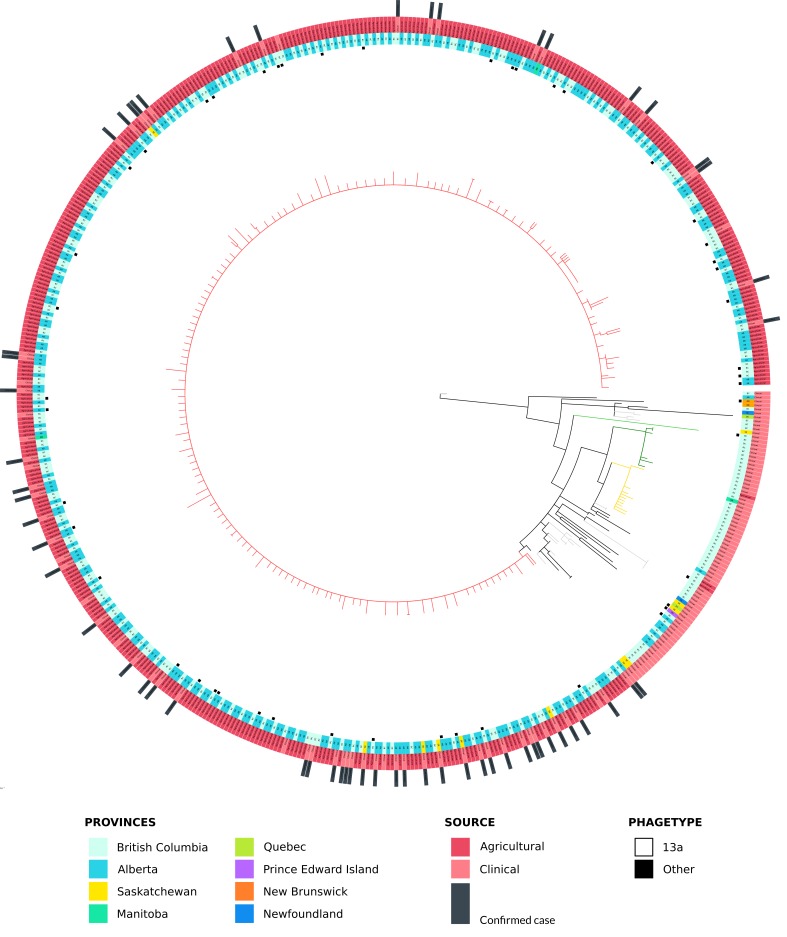
PulseNet Canada, in coordination with the affected provinces, used genome sequencing of human and agricultural Salmonella Enteritidis isolates to aid in the epidemiological investigation, while also using traditional typing methods such as phagetyping and pulsed-field gel electrophoresis (PFGE).
All human outbreak cases, except one, were Phage Type (PT) 13a. Single nucleotide variant analysis (SNV) was able to provide a level of resolution commensurate with the results of the epidemiological investigation. SNV analysis was also able to separate PT13a outbreak-related isolates from isolates not linked to chicks or poults, while clustering some non-PT13a agricultural strains with the outbreak cluster.
Based on conventional typing methods (phagetyping or PFGE), clinical and agricultural PT13a SE isolates would have been considered as part of a related cluster. In contrast, phagetyping would have led to the exclusion of several non- PT13a strains that clustered with the outbreak isolates using the genome sequence data. This study demonstrates the improved resolution of genome sequence analysis for coordinated surveillance and source attribution of both human and agricultural SE isolates.
2015年春季,一场多省范围内的肠炎沙门氏菌肠炎血清型疫情与来自单一孵化场的新孵出雏鸡和小火鸡有关。共有61例人类病例经流行病学确认与这些雏鸡和小火鸡有关,该疫情被认为在2015年夏季结束。
加拿大脉冲网与受影响省份合作,对人类和农业来源的肠炎沙门氏菌分离株进行基因组测序,以协助流行病学调查,同时也使用传统分型方法,如噬菌体分型和脉冲场凝胶电泳(PFGE)。
除1例病例外,所有人类疫情病例均为噬菌体分型(PT)13a型。单核苷酸变异分析(SNV)能够提供与流行病学调查结果相当的分辨率水平。SNV分析还能够将与PT13a疫情相关的分离株与未与雏鸡或小火鸡相关的分离株区分开来,同时将一些非PT13a农业菌株与疫情聚类。
基于传统分型方法(噬菌体分型或PFGE),临床和农业来源的PT13a肠炎沙门氏菌分离株会被视为相关聚类的一部分。相比之下,噬菌体分型会导致排除一些使用基因组序列数据与疫情分离株聚类的非PT13a菌株。本研究证明了基因组序列分析在人类和农业来源肠炎沙门氏菌分离株的协同监测和来源归因方面具有更高的分辨率。