Department of Bioinformatics, Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, 38124 Braunschweig, Germany.
Bioinformatics. 2017 Nov 1;33(21):3396-3404. doi: 10.1093/bioinformatics/btx440.
Bacterial and archaeal viruses are crucial for global biogeochemical cycles and might well be game-changing therapeutic agents in the fight against multi-resistant pathogens. Nevertheless, it is still unclear how to best use genome sequence data for a fast, universal and accurate taxonomic classification of such viruses.
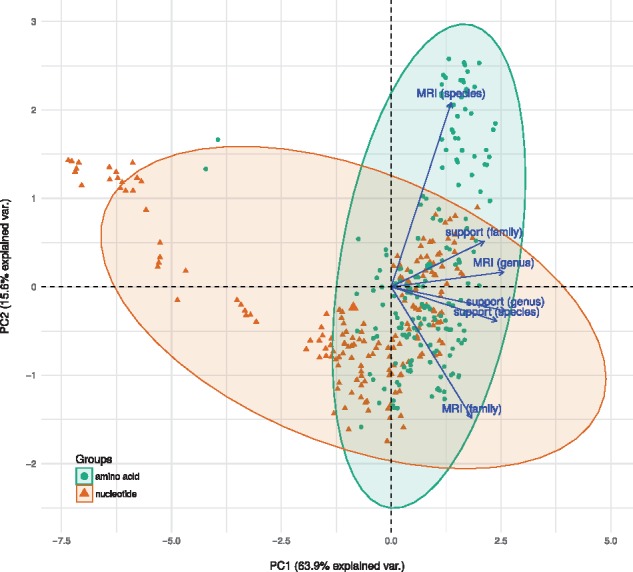
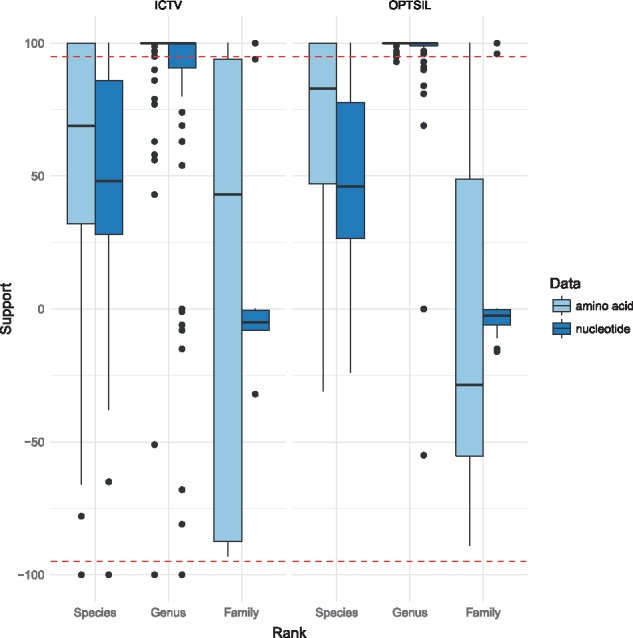
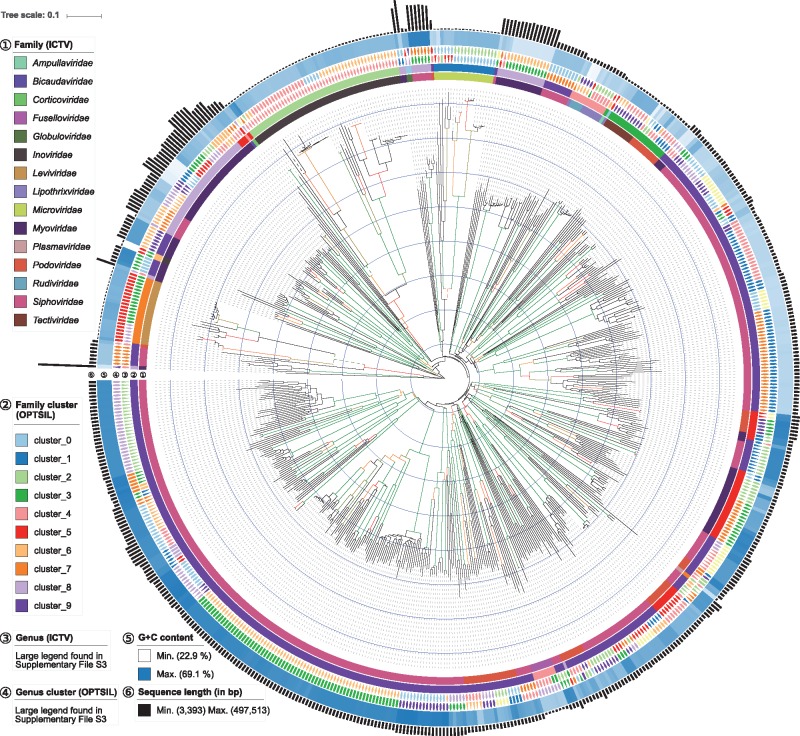
We here present a novel in silico framework for phylogeny and classification of prokaryotic viruses, in line with the principles of phylogenetic systematics, and using a large reference dataset of officially classified viruses. The resulting trees revealed a high agreement with the classification. Except for low resolution at the family level, the majority of taxa was well supported as monophyletic. Clusters obtained with distance thresholds chosen for maximizing taxonomic agreement appeared phylogenetically reasonable, too. Analysis of an expanded dataset, containing >4000 genomes from public databases, revealed a large number of novel species, genera, subfamilies and families.
The selected methods are available as the easy-to-use web service 'VICTOR' at https://victor.dsmz.de.
Supplementary data are available at Bioinformatics online.
细菌和古菌病毒对于全球生物地球化学循环至关重要,并且可能成为对抗多耐药病原体的极具变革性的治疗剂。然而,目前仍不清楚如何最好地利用基因组序列数据,快速、普遍且准确地对这些病毒进行分类。
我们在此提出了一种新的基于计算的方法,用于对原核病毒进行系统发育和分类,该方法符合系统发育分类学的原则,并使用了大量官方分类病毒的参考数据集。生成的树与分类高度一致。除了在科一级分辨率较低外,大多数分类群都支持单系性。选择最大化分类一致性的距离阈值获得的聚类在系统发育上也是合理的。对包含来自公共数据库的 >4000 个基因组的扩展数据集的分析揭示了大量新的物种、属、亚科和科。
所选方法可作为易于使用的网络服务“VICTOR”在 https://victor.dsmz.de 上使用。
补充数据可在 Bioinformatics 在线获得。