Liu Hui-Min, He Jing-Yang, Zhang Qiang, Lv Wan-Qiang, Xia Xin, Sun Chang-Qing, Zhang Wei-Dong, Deng Hong-Wen
College of Public Health Zhengzhou University, No.100 Kexue Road, High-Tech Development Zone of States, Zhengzhou, People's Republic of China.
Department of Biostatistics and Data Science, Tulane Center of Bioinformatics and Genomics, Tulane University School of Public Health and Tropical Medicine, New Orleans, LA, 70112, USA.
Mol Genet Genomics. 2018 Feb;293(1):225-235. doi: 10.1007/s00438-017-1381-6. Epub 2017 Oct 16.
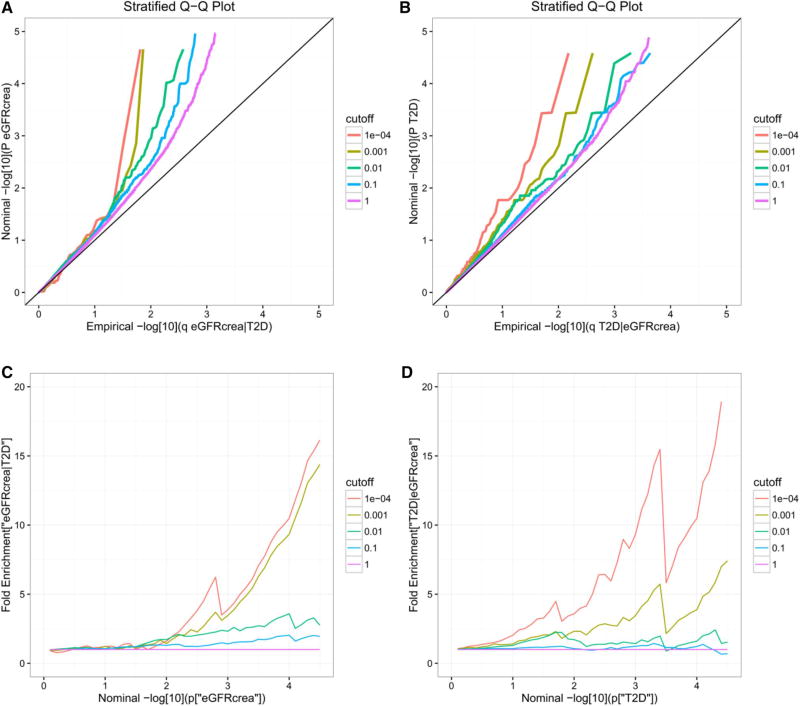
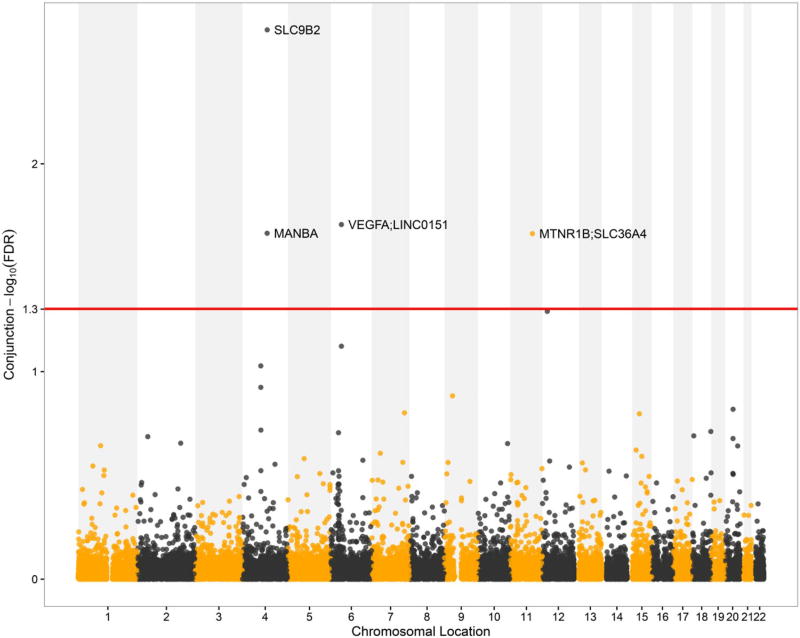
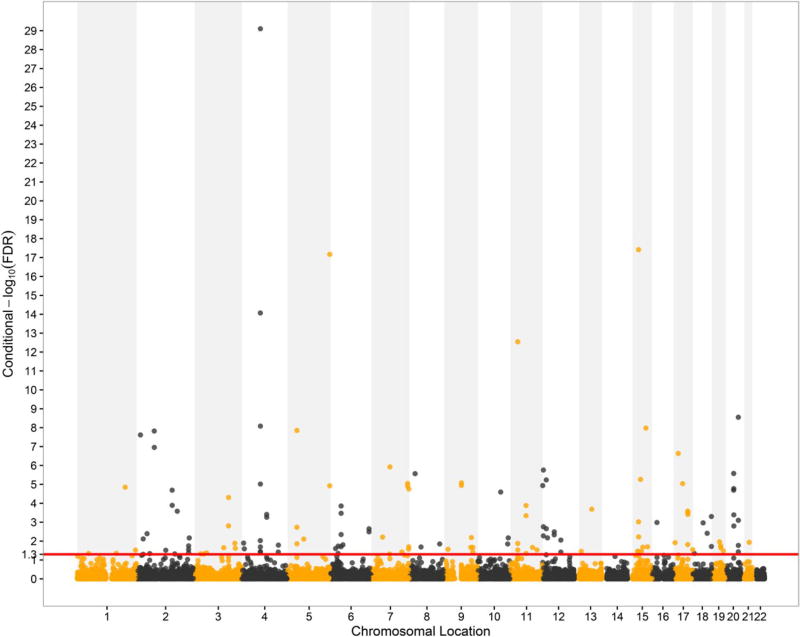
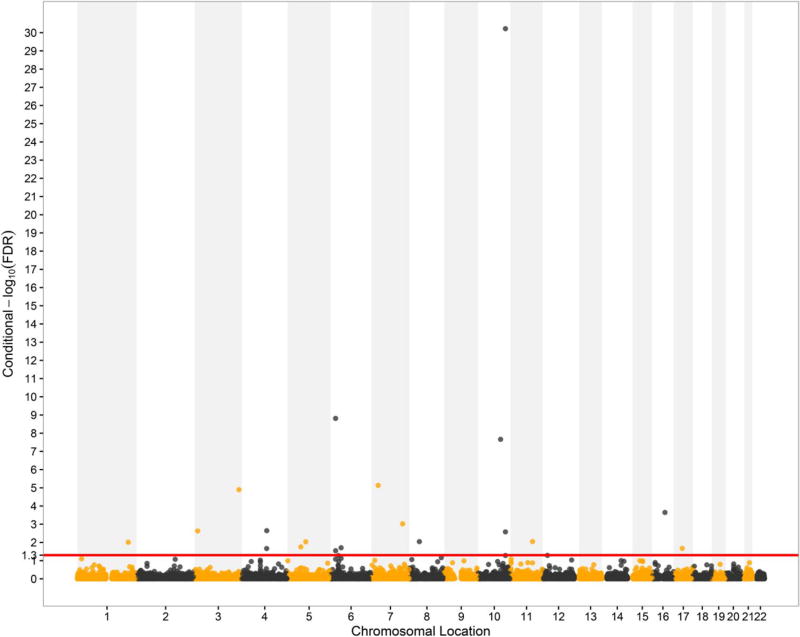
Genome-wide association studies (GWAS) have been shown to have the potential of explaining more of the "missing heritability" of complex human phenotypes by improving statistical approaches. Here, we applied a genetic-pleiotropy-informed conditional false discovery rate (cFDR) to capture additional polygenic effects associated with estimated glomerular filtration rate (creatinine) (eGFRcrea) and type 2 diabetes (T2D). The cFDR analysis improves the identification of pleiotropic variants by incorporating potentially shared genetic mechanisms between two related traits. The Q-Q and fold-enrichment plots were used to assess the enrichment of SNPs associated with eGFRcrea or T2D, and Manhattan plots were used for showing chromosomal locations of the significant loci detected. By applying the cFDR method, we newly identified 74 loci for eGFRcrea and 3 loci for T2D with the cFDR criterion of 0.05 compared with previous related GWAS studies. Four shared SNPs were detected to be associated with both eGFRcrea and T2D at the significant conjunction cFDR level of 0.05, and these shared SNPs had not been reported in previous studies. In addition, we used DAVID analysis to perform functional analysis of the shared SNPs' annotated genes and found their potential hidden associations with eGFRcrea and T2D. In this study, the cFDR method shows the feasibility to detect more genetic variants underlying the heritability of eGFRcrea and T2D, and the overlapping SNPs identified could be regarded as candidate loci that provide a thread of genetic mechanisms between eGFRcrea and T2D in future research.
全基因组关联研究(GWAS)已被证明有潜力通过改进统计方法来解释更多复杂人类表型的“缺失遗传力”。在此,我们应用了一种基于遗传多效性的条件错误发现率(cFDR)来捕获与估计肾小球滤过率(肌酐)(eGFRcrea)和2型糖尿病(T2D)相关的额外多基因效应。cFDR分析通过纳入两个相关性状之间潜在共享的遗传机制,改进了对多效性变异的识别。使用Q-Q图和富集倍数图来评估与eGFRcrea或T2D相关的单核苷酸多态性(SNP)的富集情况,并用曼哈顿图展示检测到的显著位点的染色体位置。通过应用cFDR方法,与之前相关的GWAS研究相比,我们在cFDR标准为0.05时新发现了74个与eGFRcrea相关的位点和3个与T2D相关的位点。在显著联合cFDR水平为0.05时,检测到4个共享SNP与eGFRcrea和T2D均相关,且这些共享SNP在之前的研究中未被报道。此外,我们使用DAVID分析对共享SNP的注释基因进行功能分析,发现了它们与eGFRcrea和T2D潜在的隐藏关联。在本研究中,cFDR方法显示了检测更多eGFRcrea和T2D遗传力潜在遗传变异的可行性,所识别出的重叠SNP可被视为候选位点,为未来研究中eGFRcrea和T2D之间的遗传机制提供线索。