Lin Nan, Zhu Yun, Fan Ruzong, Xiong Momiao
Department of Biostatistics and Data Science, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX, United States of America.
Department of Epidemiology, Tulane University School of Public Health and Tropical Medicine, New Orleans, LA, United States of America.
PLoS Comput Biol. 2017 Oct 17;13(10):e1005788. doi: 10.1371/journal.pcbi.1005788. eCollection 2017 Oct.
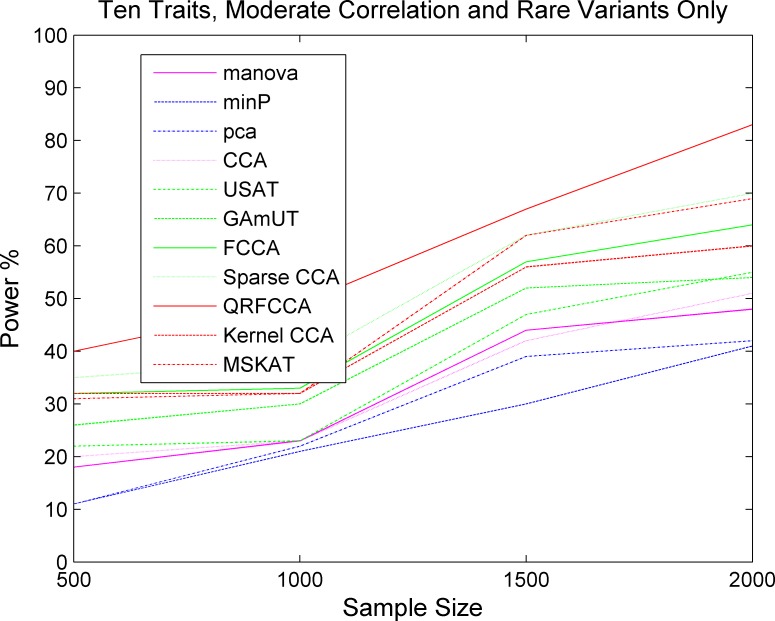
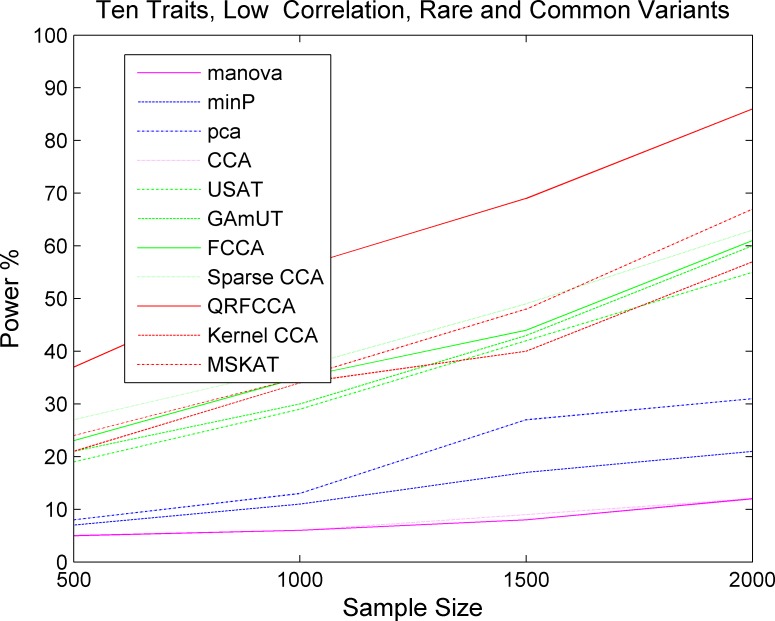
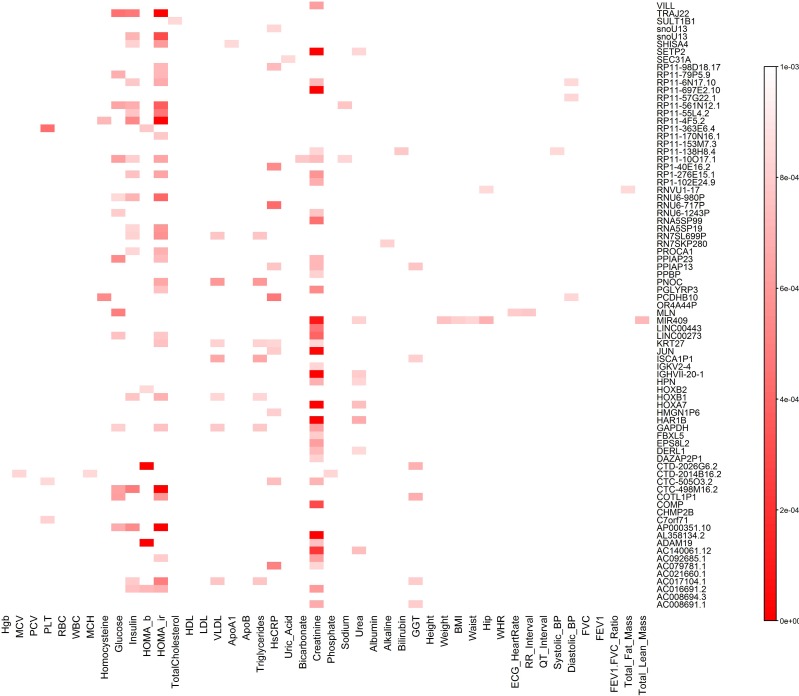
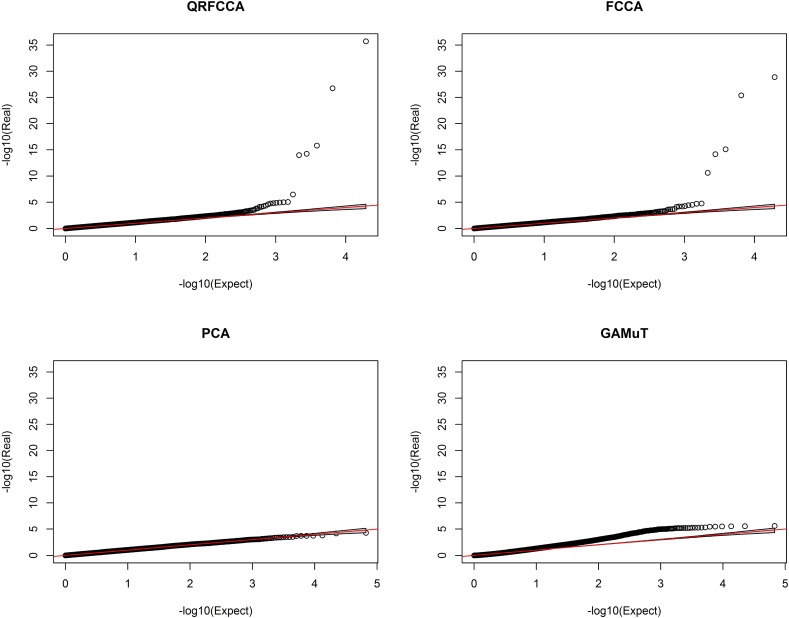
Investigating the pleiotropic effects of genetic variants can increase statistical power, provide important information to achieve deep understanding of the complex genetic structures of disease, and offer powerful tools for designing effective treatments with fewer side effects. However, the current multiple phenotype association analysis paradigm lacks breadth (number of phenotypes and genetic variants jointly analyzed at the same time) and depth (hierarchical structure of phenotype and genotypes). A key issue for high dimensional pleiotropic analysis is to effectively extract informative internal representation and features from high dimensional genotype and phenotype data. To explore correlation information of genetic variants, effectively reduce data dimensions, and overcome critical barriers in advancing the development of novel statistical methods and computational algorithms for genetic pleiotropic analysis, we proposed a new statistic method referred to as a quadratically regularized functional CCA (QRFCCA) for association analysis which combines three approaches: (1) quadratically regularized matrix factorization, (2) functional data analysis and (3) canonical correlation analysis (CCA). Large-scale simulations show that the QRFCCA has a much higher power than that of the ten competing statistics while retaining the appropriate type 1 errors. To further evaluate performance, the QRFCCA and ten other statistics are applied to the whole genome sequencing dataset from the TwinsUK study. We identify a total of 79 genes with rare variants and 67 genes with common variants significantly associated with the 46 traits using QRFCCA. The results show that the QRFCCA substantially outperforms the ten other statistics.
研究基因变异的多效性作用可以提高统计效能,为深入理解疾病复杂的遗传结构提供重要信息,并为设计副作用更少的有效治疗方法提供有力工具。然而,当前的多表型关联分析范式缺乏广度(同时联合分析的表型和基因变异数量)和深度(表型和基因型的层次结构)。高维多效性分析的一个关键问题是从高维基因型和表型数据中有效地提取信息丰富的内部表示和特征。为了探索基因变异的相关信息,有效降低数据维度,并克服推进基因多效性分析新统计方法和计算算法发展中的关键障碍,我们提出了一种新的统计方法,称为二次正则化函数典型相关分析(QRFCCA)用于关联分析,该方法结合了三种方法:(1)二次正则化矩阵分解,(2)函数数据分析和(3)典型相关分析(CCA)。大规模模拟表明,QRFCCA在保持适当的一类错误率的同时,比十种竞争统计方法具有更高的效能。为了进一步评估性能,将QRFCCA和其他十种统计方法应用于来自英国双胞胎研究的全基因组测序数据集。我们使用QRFCCA共鉴定出79个具有罕见变异的基因和67个具有常见变异且与46个性状显著相关的基因。结果表明,QRFCCA明显优于其他十种统计方法。