Department of Genetics, Stanford University, Stanford, California 94305, USA.
Department of Biology, Stanford University, Stanford, California 94305, USA.
Genome Res. 2017 Dec;27(12):1988-2000. doi: 10.1101/gr.219956.116. Epub 2017 Oct 27.
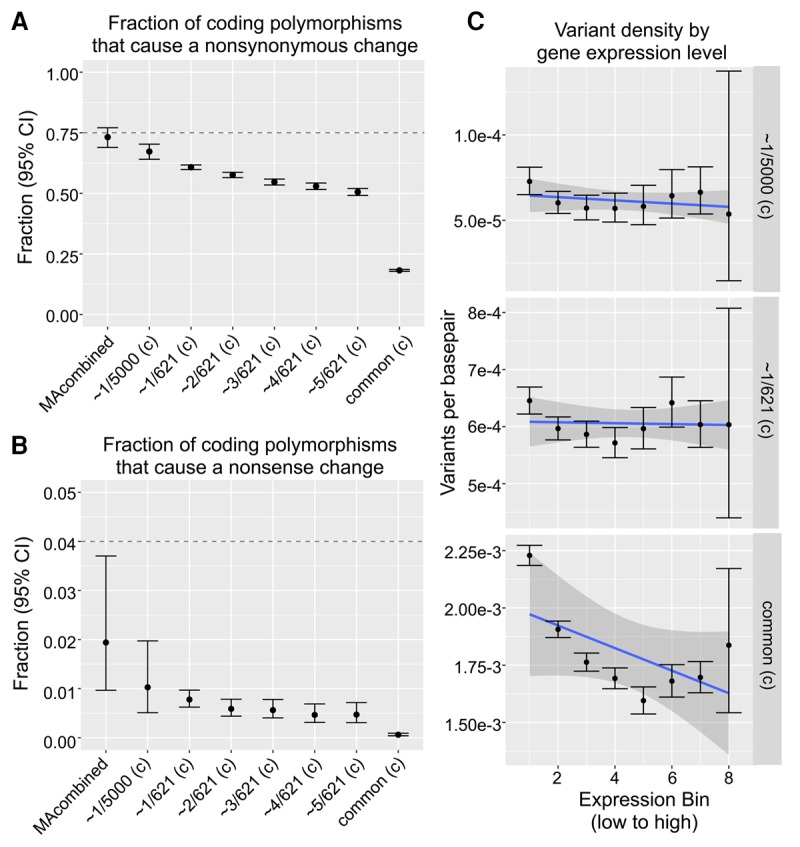
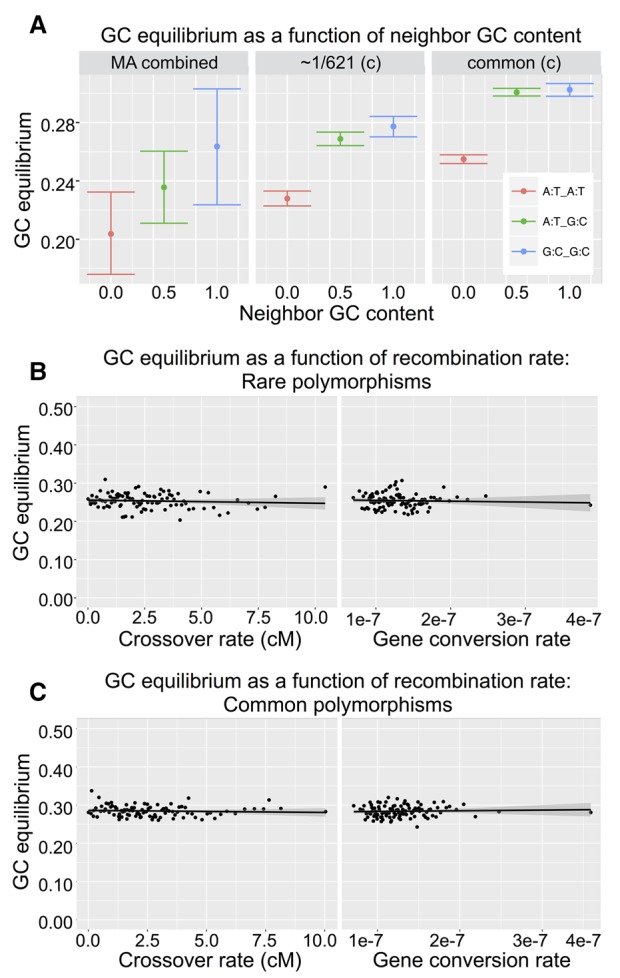
Mutations provide the raw material of evolution, and thus our ability to study evolution depends fundamentally on having precise measurements of mutational rates and patterns. We generate a data set for this purpose using (1) de novo mutations from mutation accumulation experiments and (2) extremely rare polymorphisms from natural populations. The first, mutation accumulation (MA) lines are the product of maintaining flies in tiny populations for many generations, therefore rendering natural selection ineffective and allowing new mutations to accrue in the genome. The second, rare genetic variation from natural populations allows the study of mutation because extremely rare polymorphisms are relatively unaffected by the filter of natural selection. We use both methods in , first generating our own novel data set of sequenced MA lines and performing a meta-analysis of all published MA mutations (∼2000 events) and then identifying a high quality set of ∼70,000 extremely rare (≤0.1%) polymorphisms that are fully validated with resequencing. We use these data sets to precisely measure mutational rates and patterns. Highlights of our results include: a high rate of multinucleotide mutation events at both short (∼5 bp) and long (∼1 kb) genomic distances, showing that mutation drives GC content lower in already GC-poor regions, and using our precise context-dependent mutation rates to predict long-term evolutionary patterns at synonymous sites. We also show that de novo mutations from independent MA experiments display similar patterns of single nucleotide mutation and well match the patterns of mutation found in natural populations.
突变提供了进化的原材料,因此我们研究进化的能力从根本上取决于对突变率和模式进行精确测量。为此,我们使用(1)来自突变积累实验的新突变和(2)来自自然种群的极罕见多态性生成了一个数据集。首先,突变积累(MA)系是在很小的种群中维持许多代的产物,从而使自然选择无效,并允许新的突变在基因组中积累。其次,来自自然种群的罕见遗传变异允许对突变进行研究,因为极其罕见的多态性相对不受自然选择的过滤影响。我们在 中使用了这两种方法,首先生成了我们自己的 MA 系测序的新数据集,并对所有已发表的 MA 突变(约 2000 个事件)进行了荟萃分析,然后确定了一组高质量的约 70000 个极罕见(≤0.1%)的多态性,这些多态性经过重测序完全验证。我们使用这些数据集来精确测量突变率和模式。我们的研究结果包括:在短(约 5bp)和长(约 1kb)基因组距离处的多核苷酸突变事件率都很高,表明突变使 GC 含量在已经 GC 含量低的区域进一步降低,并且使用我们精确的上下文相关突变率来预测同义位点的长期进化模式。我们还表明,来自独立 MA 实验的新突变显示出相似的单核苷酸突变模式,并且很好地匹配了自然种群中发现的突变模式。