Molecular Neurogenetics Unit, Foundation IRCCS Neurological Institute Besta, Milan, Italy.
Medical Research Council Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK.
J Med Genet. 2017 Dec;54(12):815-824. doi: 10.1136/jmedgenet-2017-104822. Epub 2017 Oct 27.
Hereditary myopathy with lactic acidosis and myopathy with deficiency of succinate dehydrogenase and aconitase are variants of a recessive disorder characterised by childhood-onset early fatigue, dyspnoea and palpitations on trivial exercise. The disease is non-progressive, but life-threatening episodes of widespread weakness, metabolic acidosis and rhabdomyolysis may occur. So far, this disease has been molecularly defined only in Swedish patients, all homozygous for a deep intronic splicing affecting mutation in encoding a scaffold protein for the assembly of iron-sulfur (Fe-S) clusters. A single Scandinavian family was identified with a different mutation, a missense change in compound heterozygosity with the common intronic mutation. The aim of the study was to identify the genetic defect in our proband.
A next-generation sequencing (NGS) approach was carried out on an Italian male who presented in childhood with ptosis, severe muscle weakness and exercise intolerance. His disease was slowly progressive, with partial recovery between episodes. Patient's specimens and yeast models were investigated.
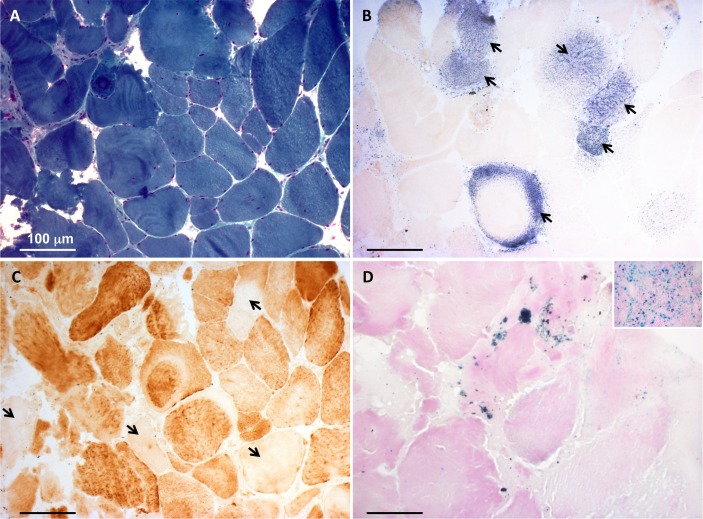
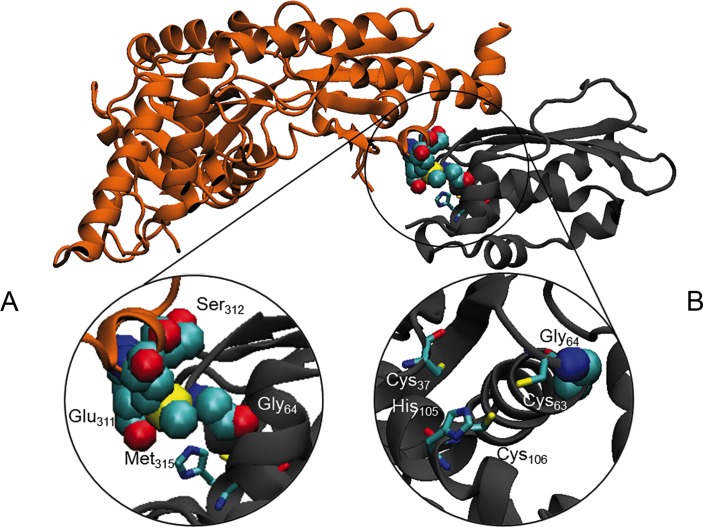
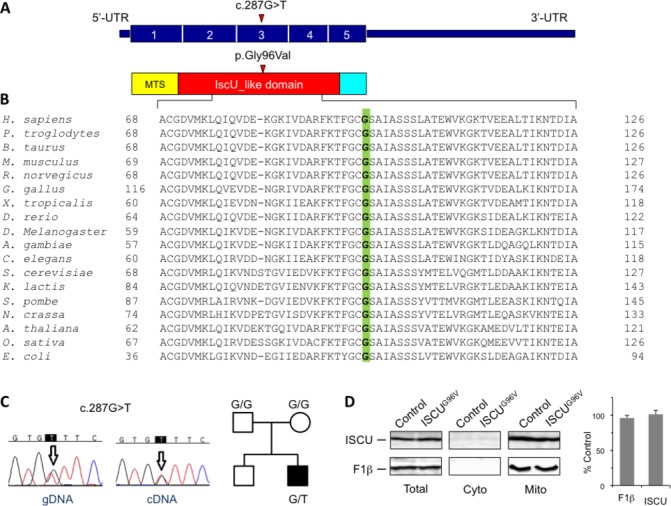
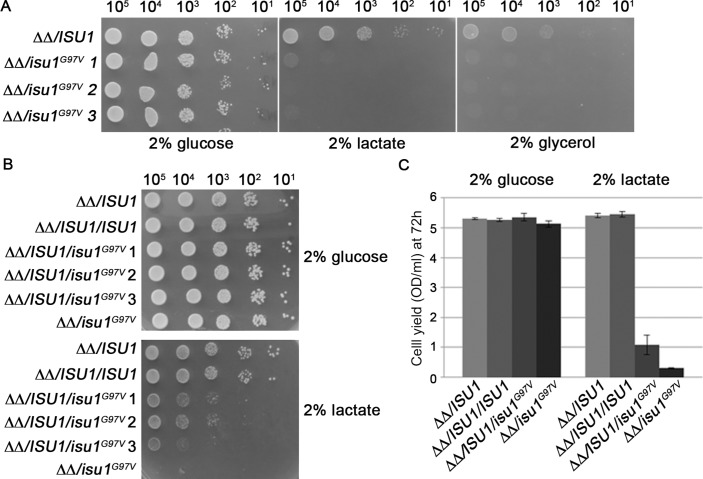
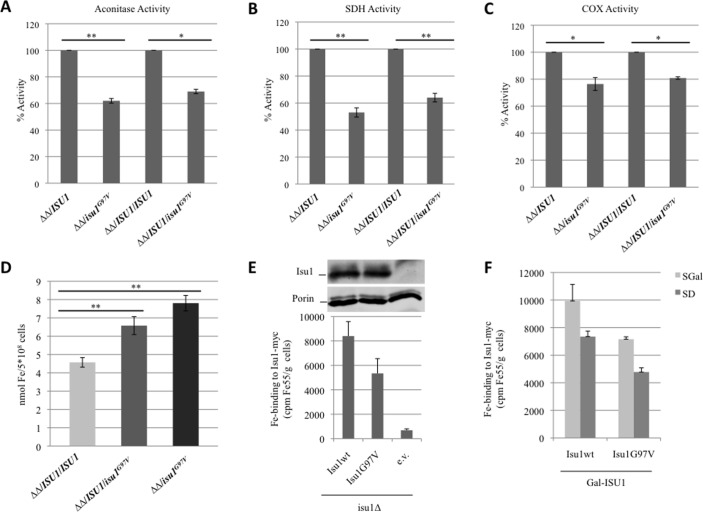
Histochemical and biochemical analyses on muscle biopsy showed multiple defects affecting mitochondrial respiratory chain complexes. We identified a single heterozygous mutation p.Gly96Val in , which was absent in DNA from his parents indicating a possible de novo dominant effect in the patient. Patient fibroblasts showed normal levels of ISCU protein and a few variably affected Fe-S cluster-dependent enzymes. Yeast studies confirmed both pathogenicity and dominance of the identified missense mutation.
We describe the first heterozygous dominant mutation in which results in a phenotype reminiscent of the recessive disease previously reported.
遗传性肌病伴乳酸性酸中毒和琥珀酸脱氢酶及顺乌头酸酶缺乏性肌病是一种隐性疾病的变异,其特征为儿童期起病,轻微运动即出现早期疲劳、呼吸困难和心悸。该疾病是非进行性的,但可能发生广泛无力、代谢性酸中毒和横纹肌溶解等危及生命的发作。迄今为止,这种疾病仅在瑞典患者中进行了分子定义,所有患者均为影响 编码铁硫(Fe-S)簇组装支架蛋白的深内含子剪接突变的纯合子。在一个不同的突变中,在一个具有共同内含子突变的复合杂合子中,确定了一个单一的斯堪的纳维亚家族。本研究的目的是确定我们的先证者的遗传缺陷。
对一名意大利男性进行下一代测序(NGS),该男性在儿童时期出现上睑下垂、严重肌肉无力和运动不耐受。他的疾病进展缓慢,发作之间有部分恢复。对患者的标本和酵母模型进行了研究。
肌肉活检的组织化学和生化分析显示,多个缺陷影响线粒体呼吸链复合物。我们在 中发现了一个单一的杂合突变 p.Gly96Val,其在患者父母的 DNA 中不存在,表明患者可能存在新的显性效应。患者的成纤维细胞显示出正常水平的 ISCU 蛋白和一些可变影响的 Fe-S 簇依赖性酶。酵母研究证实了所鉴定的错义突变的致病性和显性。
我们描述了 中的第一个杂合显性突变,其表型类似于先前报道的隐性疾病。