Department of Cardiology, Shanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai, China.
Department of Psychiatry, Centre for Genomic Sciences, The University of Hong Kong, Pokfulam, Hong Kong.
J Am Heart Assoc. 2017 Oct 28;6(11):e007030. doi: 10.1161/JAHA.117.007030.
There is increasing interest in the concept of atrial cardiomyopathy, but the underlying molecular and mechanistic determinants remain poorly defined. We identified a family with heritable atrial cardiomyopathy manifesting as progressive atrial-selective electromechanical dysfunction, tachyarrhythmias, and bradyarrhythmias requiring pacemaker implantation. Myosin light-chain 4 (), encoding the atrial-selective essential myosin light chain, was identified as a candidate gene. We used genetically modified rat models to investigate the role of in atrial cardiomyopathy.
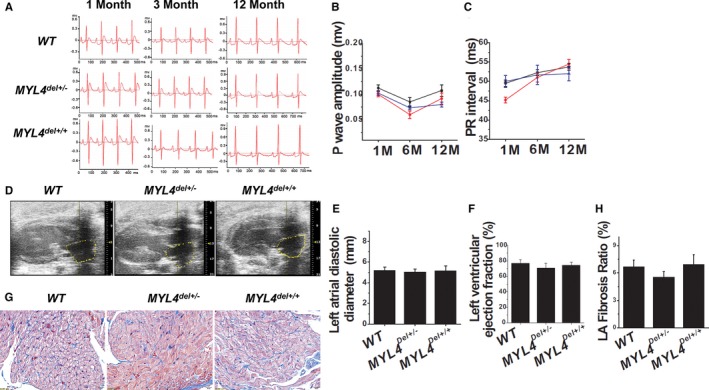
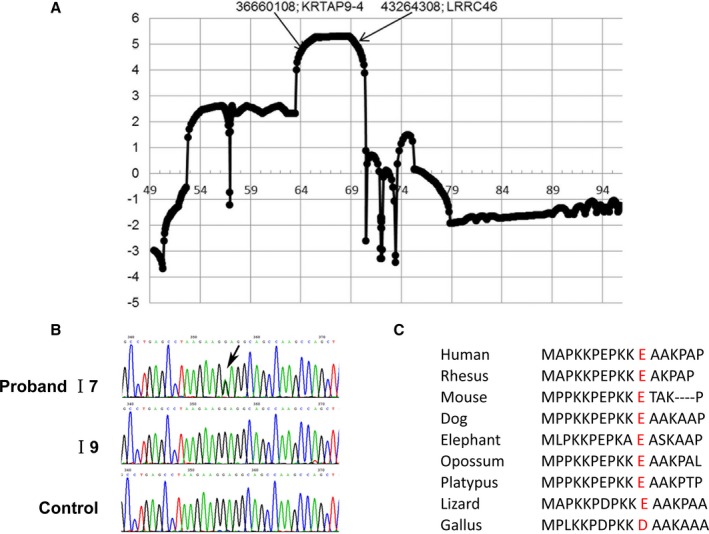
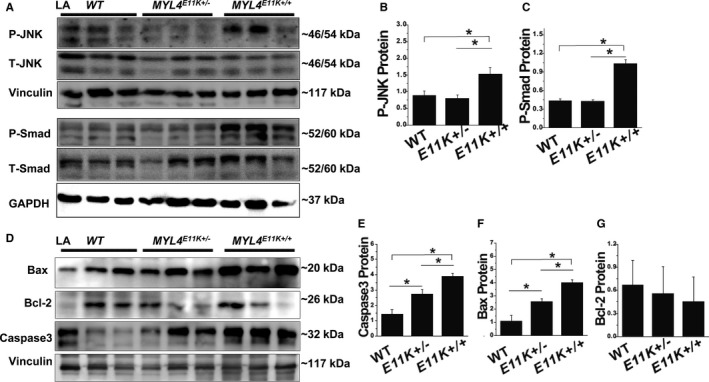
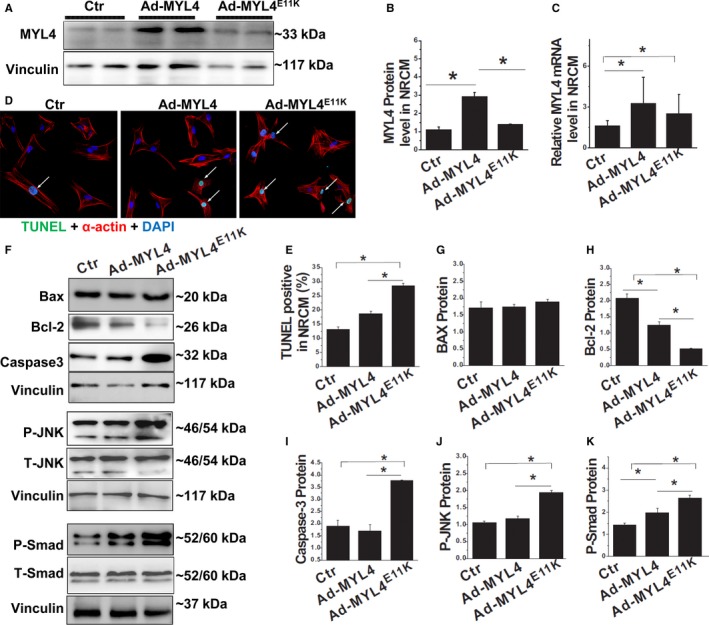
Exome sequencing and systematic bioinformatic analyses identified a rare missense variant of (c.31G>A []) in a large multiplex atrial cardiomyopathy family pedigree. The mutation cosegregated with atrial standstill (selected as the principal presenting trait) with a logarithm of the odds score of 5.3. The phenotype of rats with mutation knock-in confirmed the causative role of the mutation. knockout rats showed a similar atrial cardiomyopathy phenotype, whereas rats with an adjacent 4-amino-acid deletion showed no phenotype. Both knock-in rats and knockout rats showed progressive atrial electrophysiological, contractile, and fibrotic abnormalities, similar to affected patients. Biochemical analyses of mutation rats showed activation of proapoptotic and profibrotic signaling, along with increased atrial-cardiomyocyte terminal deoxynucleotidyl transferase dUTP nick end labeling staining, suggesting enhanced apoptotic cell death, findings that were mimicked by in vitro adenoviral transfer of the mutant gene to neonatal-rat cardiomyocytes.
Loss-of-function gene variants cause progressive atrial cardiomyopathy in humans and rats. Our findings identify as a key gene required for atrial contractile, electrical and structural integrity. These results improve our understanding of the molecular basis of atrial cardiomyopathy and introduce new models for further mechanistic analysis.
人们对心房心肌病的概念越来越感兴趣,但潜在的分子和机制决定因素仍未得到明确界定。我们发现了一个家族,其遗传性心房心肌病表现为进行性心房选择性机电功能障碍、心动过速性心律失常和心动过缓性心律失常,需要植入起搏器。肌球蛋白轻链 4(),编码心房选择性必需肌球蛋白轻链,被鉴定为候选基因。我们使用基因修饰大鼠模型来研究在心房心肌病中的作用。
外显子组测序和系统生物信息学分析在一个大型多房性心肌病家族系谱中发现了一个罕见的肌球蛋白轻链 4()错义变异(c.31G>A [])。该突变与心房停搏(作为主要表现特征选择)共分离,对数优势评分为 5.3。突变敲入大鼠的表型证实了该突变的因果作用。肌球蛋白轻链 4 基因敲除大鼠表现出类似的心房心肌病表型,而相邻的 4 个氨基酸缺失大鼠则没有表型。肌球蛋白轻链 4 敲入大鼠和肌球蛋白轻链 4 基因敲除大鼠均表现出进行性心房电生理、收缩和纤维化异常,与受影响的患者相似。肌球蛋白轻链 4 突变大鼠的生化分析显示促凋亡和促纤维化信号的激活,以及心房心肌细胞末端脱氧核苷酸转移酶 dUTP 缺口末端标记染色增加,提示凋亡细胞死亡增加,这一发现通过将突变基因的腺病毒体外转染到新生大鼠心肌细胞中得到模拟。
功能丧失的肌球蛋白轻链 4 基因突变导致人类和大鼠进行性心房心肌病。我们的发现确定肌球蛋白轻链 4 为心房收缩、电和结构完整性所必需的关键基因。这些结果提高了我们对心房心肌病分子基础的理解,并引入了新的模型以进行进一步的机制分析。