Human Genetics Center, University of Texas Health Science Center, Houston, TX, USA.
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Genome Med. 2017 Oct 31;9(1):95. doi: 10.1186/s13073-017-0482-5.
Left-sided lesions (LSLs) account for an important fraction of severe congenital cardiovascular malformations (CVMs). The genetic contributions to LSLs are complex, and the mutations that cause these malformations span several diverse biological signaling pathways: TGFB, NOTCH, SHH, and more. Here, we use whole exome sequence data generated in 342 LSL cases to identify likely damaging variants in putative candidate CVM genes.
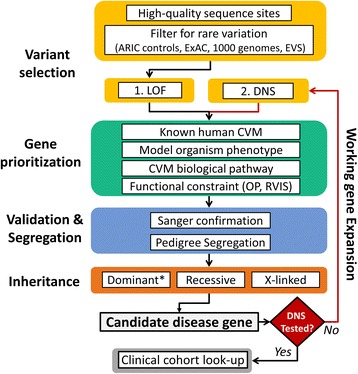
Using a series of bioinformatics filters, we focused on genes harboring population-rare, putative loss-of-function (LOF), and predicted damaging variants in 1760 CVM candidate genes constructed a priori from the literature and model organism databases. Gene variants that were not observed in a comparably sequenced control dataset of 5492 samples without severe CVM were then subjected to targeted validation in cases and parents. Whole exome sequencing data from 4593 individuals referred for clinical sequencing were used to bolster evidence for the role of candidate genes in CVMs and LSLs.
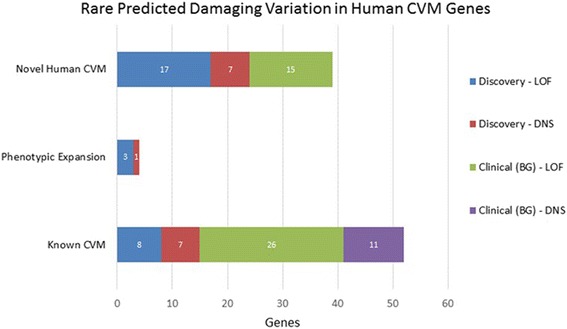
Our analyses revealed 28 candidate variants in 27 genes, including 17 genes not previously associated with a human CVM disorder, and revealed diverse patterns of inheritance among LOF carriers, including 9 confirmed de novo variants in both novel and newly described human CVM candidate genes (ACVR1, JARID2, NR2F2, PLRG1, SMURF1) as well as established syndromic CVM genes (KMT2D, NF1, TBX20, ZEB2). We also identified two genes (DNAH5, OFD1) with evidence of recessive and hemizygous inheritance patterns, respectively. Within our clinical cohort, we also observed heterozygous LOF variants in JARID2 and SMAD1 in individuals with cardiac phenotypes, and collectively, carriers of LOF variants in our candidate genes had a four times higher odds of having CVM (odds ratio = 4.0, 95% confidence interval 2.5-6.5).
Our analytical strategy highlights the utility of bioinformatic resources, including human disease records and model organism phenotyping, in novel gene discovery for rare human disease. The results underscore the extensive genetic heterogeneity underlying non-syndromic LSLs, and posit potential novel candidate genes and complex modes of inheritance in this important group of birth defects.
左侧病变(LSL)占严重先天性心血管畸形(CVM)的重要部分。导致这些畸形的遗传因素很复杂,引起这些畸形的突变跨越了几个不同的生物信号通路:TGFB、NOTCH、SHH 等。在这里,我们使用在 342 例 LSL 病例中生成的全外显子组序列数据,来识别假定的候选 CVM 基因中的可能有害变异。
使用一系列生物信息学筛选,我们专注于在 1760 个候选 CVM 基因中,那些携带人群罕见的、潜在的功能丧失(LOF)和预测有害变异的基因。在没有严重 CVM 的 5492 个样本的对照测序数据集中未观察到的基因变体,然后在病例和父母中进行靶向验证。对 4593 名接受临床测序的个体的全外显子组测序数据进行了使用,以支持候选基因在 CVM 和 LSL 中的作用的证据。
我们的分析揭示了 27 个基因中的 28 个候选变异,包括 17 个以前与人类 CVM 疾病无关的基因,并且在 LOF 携带者中显示出不同的遗传模式,包括在新的和新描述的人类 CVM 候选基因(ACVR1、JARID2、NR2F2、PLRG1、SMURF1)中以及已建立的综合征性 CVM 基因(KMT2D、NF1、TBX20、ZEB2)中确认的 9 个新生的和新描述的 CVM 候选基因(ACVR1、JARID2、NR2F2、PLRG1、SMURF1)以及已建立的综合征性 CVM 基因(KMT2D、NF1、TBX20、ZEB2)中确认的 de novo 变异。我们还分别在 DNAH5 和 OFD1 基因中发现了证据确凿的隐性和半合子遗传模式。在我们的临床队列中,我们还观察到 JARID2 和 SMAD1 基因的杂合性 LOF 变异存在于有心脏表型的个体中,并且在我们的候选基因中携带 LOF 变异的携带者患 CVM 的可能性高出四倍(比值比=4.0,95%置信区间 2.5-6.5)。
我们的分析策略突出了生物信息学资源的实用性,包括人类疾病记录和模式生物表型,在罕见人类疾病的新基因发现中。结果强调了非综合征性 LSL 背后广泛的遗传异质性,并提出了该重要出生缺陷群体中的潜在新候选基因和复杂遗传模式。