Kneynsberg Andrew, Combs Benjamin, Christensen Kyle, Morfini Gerardo, Kanaan Nicholas M
Neuroscience Program, Michigan State University, East Lansing, MI, United States.
Department of Translational Science and Molecular Medicine, College of Human Medicine, Michigan State University, Grand Rapids, MI, United States.
Front Neurosci. 2017 Oct 17;11:572. doi: 10.3389/fnins.2017.00572. eCollection 2017.
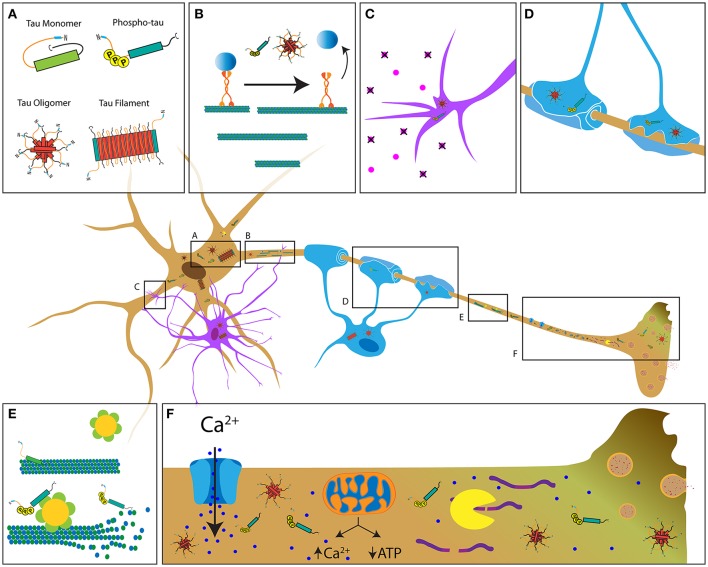
Tauopathies are a diverse group of diseases featuring progressive dying-back neurodegeneration of specific neuronal populations in association with accumulation of abnormal forms of the microtubule-associated protein tau. It is well-established that the clinical symptoms characteristic of tauopathies correlate with deficits in synaptic function and neuritic connectivity early in the course of disease, but mechanisms underlying these critical pathogenic events are not fully understood. Biochemical evidence fueled the widespread notion that microtubule stabilization represents tau's primary biological role and that the marked atrophy of neurites observed in tauopathies results from loss of microtubule stability. However, this notion contrasts with the mild phenotype associated with tau deletion. Instead, an analysis of cellular hallmarks common to different tauopathies, including aberrant patterns of protein phosphorylation and early degeneration of axons, suggests that alterations in kinase-based signaling pathways and deficits in axonal transport (AT) associated with such alterations contribute to the loss of neuronal connectivity triggered by pathogenic forms of tau. Here, we review a body of literature providing evidence that axonal pathology represents an early and common pathogenic event among human tauopathies. Observations of axonal degeneration in animal models of specific tauopathies are discussed and similarities to human disease highlighted. Finally, we discuss potential mechanistic pathways other than microtubule destabilization by which disease-related forms of tau may promote axonopathy.
tau蛋白病是一类多样的疾病,其特征是特定神经元群体进行性逆行性神经变性,并伴有微管相关蛋白tau异常形式的积累。众所周知,tau蛋白病的临床症状与疾病早期突触功能和神经突连接缺陷相关,但这些关键致病事件的潜在机制尚未完全了解。生化证据支持了一种广泛的观点,即微管稳定是tau蛋白的主要生物学作用,并且在tau蛋白病中观察到的神经突明显萎缩是由于微管稳定性丧失所致。然而,这一观点与tau蛋白缺失相关的轻度表型形成对比。相反,对不同tau蛋白病共有的细胞特征进行分析,包括异常的蛋白磷酸化模式和轴突早期变性,表明基于激酶的信号通路改变以及与此类改变相关的轴突运输(AT)缺陷,导致了由致病形式的tau蛋白引发的神经元连接丧失。在此,我们回顾了一系列文献,这些文献提供了证据表明轴突病理学是人类tau蛋白病中早期且常见的致病事件。讨论了特定tau蛋白病动物模型中轴突变性的观察结果,并强调了与人类疾病的相似性。最后,我们讨论了除微管去稳定化之外的潜在机制途径,疾病相关形式的tau蛋白可能通过这些途径促进轴突病变。