Thiffault Isabelle, Zuccarelli Britton, Welsh Holly, Yuan Xuan, Farrow Emily, Zellmer Lee, Miller Neil, Soden Sarah, Abdelmoity Ahmed, Brodsky Robert A, Saunders Carol
Center for Pediatric Genomic Medicine, Children's Mercy Hospital, 2420 Pershing Road, Kansas City, MO, 64108, USA.
Department of Pathology and Laboratory Medicine, Children's Mercy Hospitals, Kansas City, MO, USA.
BMC Med Genet. 2017 Nov 2;18(1):124. doi: 10.1186/s12881-017-0481-9.
Defects in the human glycosylphosphatidylinositol anchor biosynthetic pathway are associated with inherited glycosylphosphatidylinositol (GPI)-deficiencies characterized by a broad range of clinical phenotypes including multiple congenital anomalies, dysmorphic faces, developmental delay, hypotonia, and epilepsy. Biallelic variants in PIGN, encoding phosphatidylinositol-glycan biosynthesis class N have been recently associated with multiple congenital anomalies hypotonia seizure syndrome.
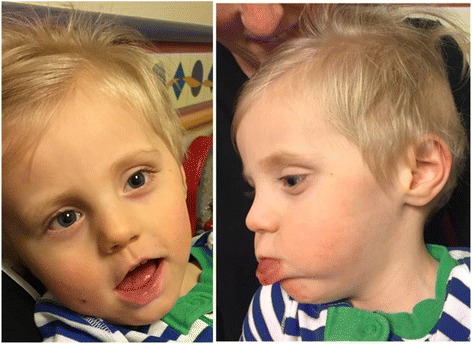
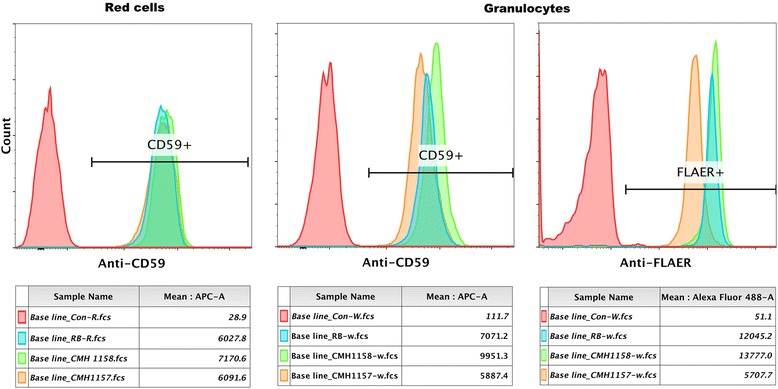
Our patient is a 2 year old male with hypotonia, global developmental delay, and focal epilepsy. Trio whole-exome sequencing revealed heterozygous variants in PIGN, c.181G > T (p.Glu61*) and c.284G > A (p.Arg95Gln). Analysis of FLAER and anti-CD59 by flow-cytometry demonstrated a shift in this patient's granulocytes, confirming a glycosylphosphatidylinositol-biosynthesis defect, consistent with PIGN-related disease.
To date, a total of 18 patients have been reported, all but 2 of whom have congenital anomalies and/or obvious dysmorphic features. Our patient has no significant dysmorphic features or multiple congenital anomalies, which is consistent with recent reports linking non-truncating variants with a milder phenotype, highlighting the importance of functional studies in interpreting sequence variants.
人类糖基磷脂酰肌醇锚生物合成途径缺陷与遗传性糖基磷脂酰肌醇(GPI)缺乏症相关,其特征为广泛的临床表型,包括多种先天性畸形、面容畸形、发育迟缓、肌张力减退和癫痫。编码磷脂酰肌醇聚糖生物合成N类的PIGN基因双等位基因变异最近与多种先天性畸形肌张力减退癫痫综合征相关。
我们的患者是一名2岁男性,有肌张力减退、全面发育迟缓及局灶性癫痫。三联体全外显子测序显示PIGN基因存在杂合变异,即c.181G>T(p.Glu61*)和c.284G>A(p.Arg95Gln)。通过流式细胞术分析FLAER和抗CD59,证实该患者粒细胞发生改变,确认存在糖基磷脂酰肌醇生物合成缺陷,与PIGN相关疾病一致。
迄今为止,共报告了18例患者,其中除2例患者外均有先天性畸形和/或明显的畸形特征。我们的患者没有明显的畸形特征或多种先天性畸形,这与最近将非截短变异与较轻表型相关联的报道一致,突出了功能研究在解释序列变异中的重要性。