Xiao Shi-Qi, Li Mei-Hui, Meng Yi-Lin, Li Chuang, Huang Hai-Long, Liu Cai-Xia, Lyu Yuan, Na Quan
Department of Nursing, Shengjing Hospital of China Medical University, Shenyang, China.
Department of Obstetrics and Gynecology, Shengjing Hospital of China Medical University, Shenyang, China.
Front Genet. 2020 Oct 27;11:594078. doi: 10.3389/fgene.2020.594078. eCollection 2020.
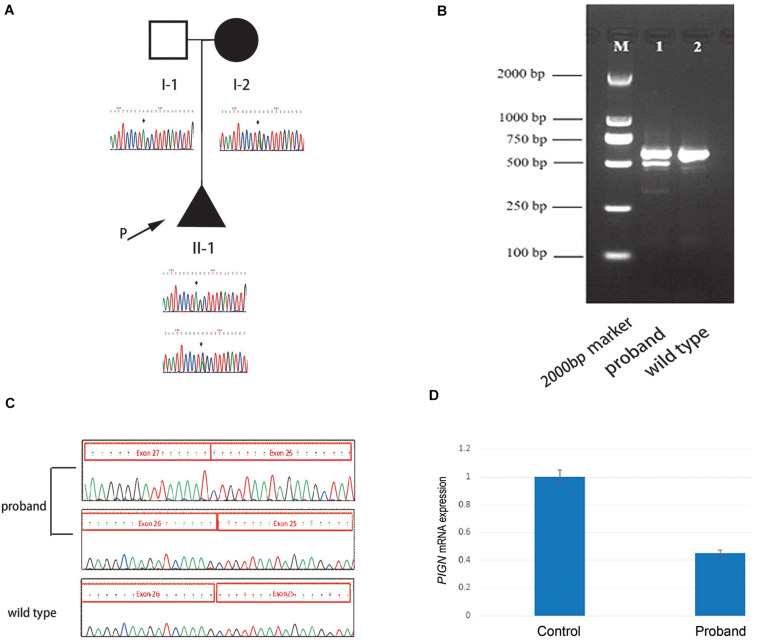
Multiple congenital anomalies-hypotonia-seizures syndrome 1 (MCAHS1) caused by phosphatidylinositol-glycan biosynthesis class N ( mutations is an autosomal recessive disease involving many systems of the body, such as the urogenital, cardiovascular, gastrointestinal, and central nervous systems. Here, compound heterozygous variants NM_012327.6:c.2427-2A > G and c.963G > A in were identified in a Chinese proband with MCAHS1. The features of the MCAHS1 family proband were evaluated to understand the mechanism of the mutation leading to the occurrence of MCAHS1. Ultrasound was conducted to examine the fetus, and his clinical manifestations were evaluated. Genetic testing was performed by whole-exome sequencing and the results were verified by Sanger sequencing of the proband and his parents. Reverse transcription-polymerase chain reaction was performed, and the products were subjected to Sanger sequencing. Quantitative PCR (Q-PCR) was conducted to compare gene expression between the patient and wild-type subjects. The compound heterozygous mutation NM_012327.6:c.2427-2A > G and c.963G > A was identified by whole-exome sequencing and was confirmed by Sanger sequencing. The NM_012327.6:c.2427-2A > G mutation led to skipping of exon 26, which resulted in a low expression level of the gene, as measured by Q-PCR. These findings provided a basis for genetic counseling and reproduction guidance in this family. Phenotype-genotype correlations may be defined by an expanded array of mutations.
由磷脂酰肌醇聚糖生物合成N类(PIG - N)基因突变引起的多发性先天性异常-肌张力减退-癫痫综合征1(MCAHS1)是一种常染色体隐性疾病,累及身体的许多系统,如泌尿生殖系统、心血管系统、胃肠道和中枢神经系统。在此,在中国一名患有MCAHS1的先证者中鉴定出复合杂合变异NM_012327.6:c.2427 - 2A > G和c.963G > A。对MCAHS1家系先证者的特征进行评估,以了解该突变导致MCAHS1发生的机制。对胎儿进行超声检查,并评估其临床表现。通过全外显子组测序进行基因检测,结果通过对先证者及其父母的Sanger测序进行验证。进行逆转录-聚合酶链反应,并对产物进行Sanger测序。进行定量PCR(Q - PCR)以比较患者与野生型受试者之间的基因表达。通过全外显子组测序鉴定出复合杂合突变NM_012327.6:c.2427 - 2A > G和c.963G > A,并通过Sanger测序得到证实。NM_012327.6:c.2427 - 2A > G突变导致外显子26跳跃,通过Q - PCR检测,这导致该基因表达水平较低。这些发现为该家族的遗传咨询和生育指导提供了依据。表型-基因型相关性可能由一系列扩展的突变来定义。