Center for Hemochromatosis, Department of Internal Medicine II, University of Modena and Reggio Emilia Policlinico, Modena, Italy
Haematologica. 2017 Dec;102(12):1972-1984. doi: 10.3324/haematol.2017.170720. Epub 2017 Nov 3.
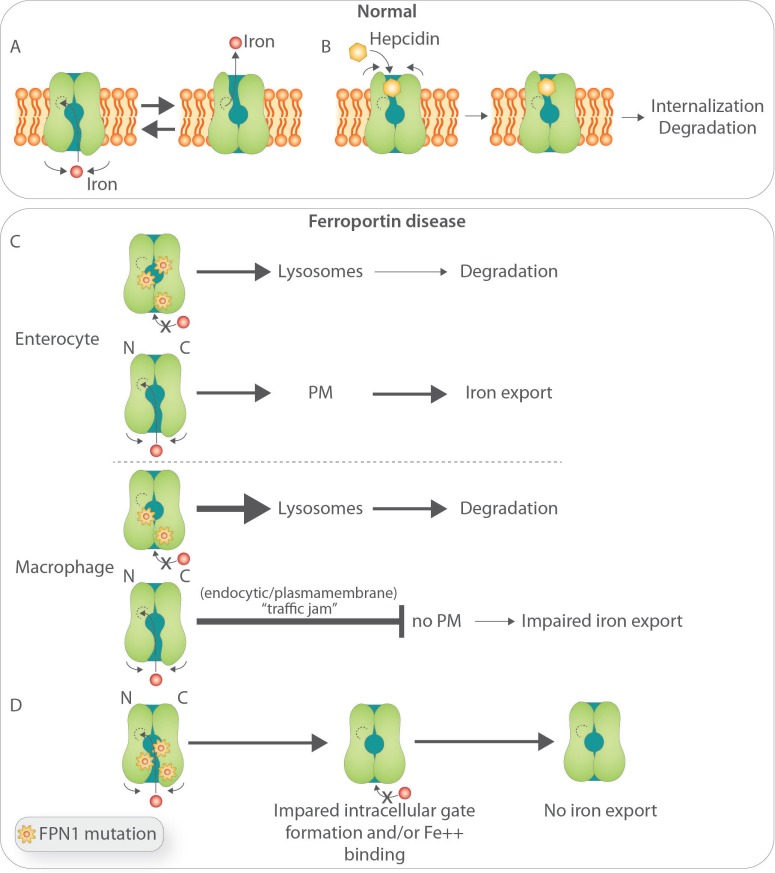
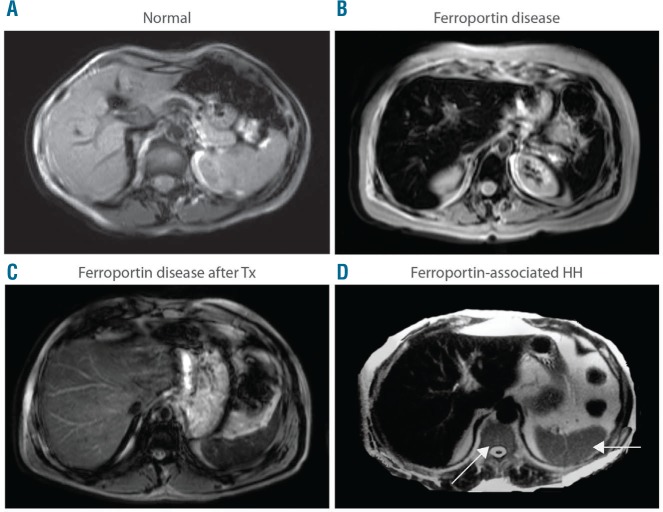
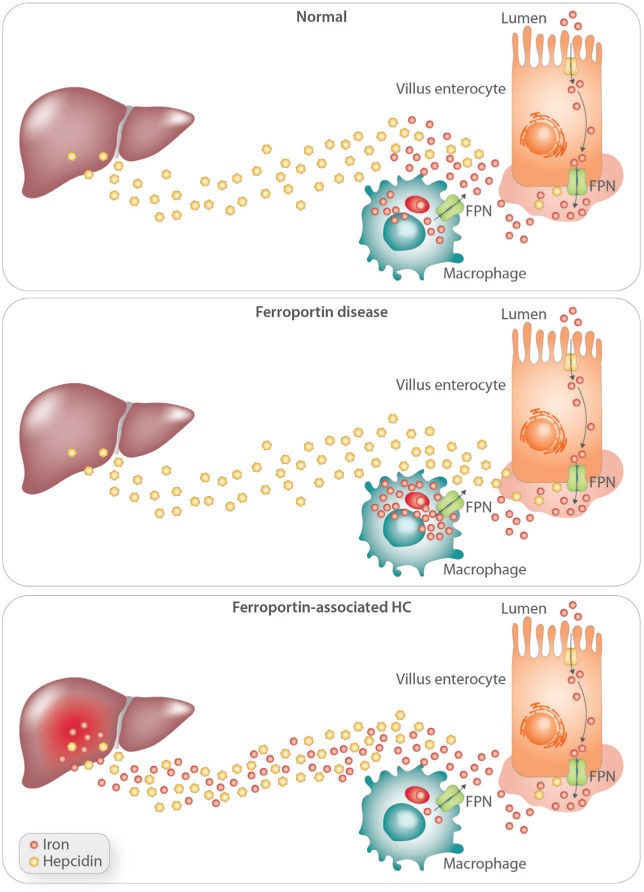
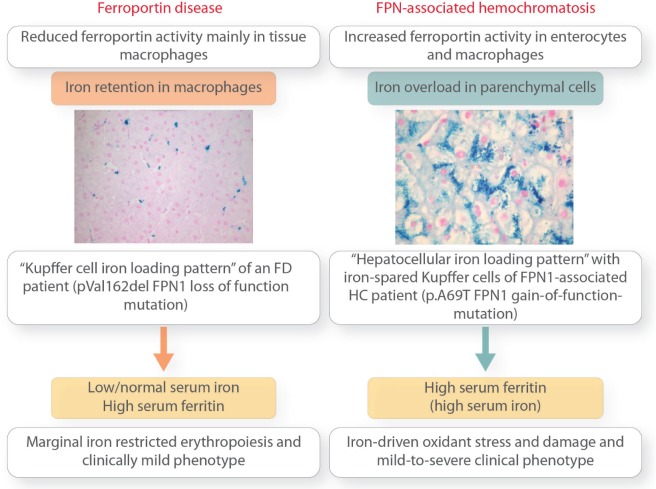
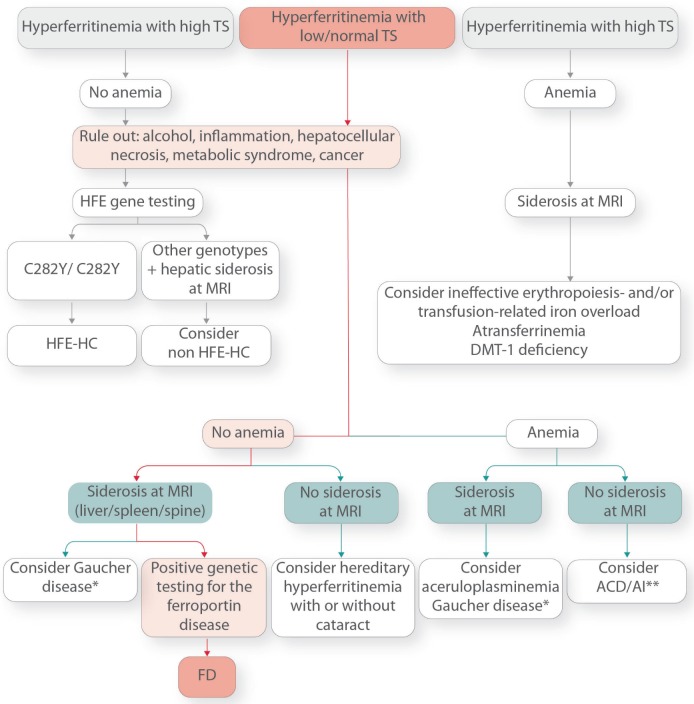
Ferroportin Disease (FD) is an autosomal dominant hereditary iron loading disorder associated with heterozygote mutations of the ferroportin-1 () gene. It represents one of the commonest causes of genetic hyperferritinemia, regardless of ethnicity. FPN1 transfers iron from the intestine, macrophages and placenta into the bloodstream. In FD, loss-of-function mutations of FPN1 limit but do not impair iron export in enterocytes, but they do severely affect iron transfer in macrophages. This leads to progressive and preferential iron trapping in tissue macrophages, reduced iron release to serum transferrin (i.e. inappropriately low transferrin saturation) and a tendency towards anemia at menarche or after intense bloodletting. The hallmark of FD is marked iron accumulation in hepatic Kupffer cells. Numerous FD-associated mutations have been reported worldwide, with a few occurring in different populations and some more commonly reported (e.g. Val192del, A77D, and G80S). FPN1 polymorphisms also represent the gene variants most commonly responsible for hyperferritinemia in Africans. Differential diagnosis includes mainly hereditary hemochromatosis, the syndrome commonly due to either or , , , and, in rare instances, itself. Here, unlike FD, hyperferritinemia associates with high transferrin saturation, iron-spared macrophages, and progressive parenchymal cell iron load. Abdominal magnetic resonance imaging (MRI), the key non-invasive diagnostic tool for the diagnosis of FD, shows the characteristic iron loading SSL triad (spleen, spine and liver). A non-aggressive phlebotomy regimen is recommended, with careful monitoring of transferrin saturation and hemoglobin due to the risk of anemia. Family screening is mandatory since siblings and offspring have a 50% chance of carrying the pathogenic mutation.
铁蛋白病(FD)是一种常染色体显性遗传性铁过载疾病,与铁蛋白-1(FPN1)基因的杂合突变有关。无论种族如何,它都是遗传性高血铁蛋白症最常见的原因之一。FPN1 将铁从肠道、巨噬细胞和胎盘转运到血液中。在 FD 中,FPN1 的功能丧失突变限制但不损害肠细胞中的铁输出,但严重影响巨噬细胞中的铁转运。这导致组织巨噬细胞中进行性和优先铁捕获,血清转铁蛋白中铁释放减少(即转铁蛋白饱和度不合适低)和初潮或大量出血后贫血的趋势。FD 的标志是肝脏枯否细胞中明显的铁积累。全世界已经报道了许多与 FD 相关的突变,其中一些在不同人群中发生,有些则更常见(例如 Val192del、A77D 和 G80S)。FPN1 多态性也代表最常见导致非洲人高血铁蛋白的基因变异。鉴别诊断主要包括遗传性血色素沉着症,该综合征通常由 或 、 、 、 引起,在极少数情况下,也可能由 本身引起。与 FD 不同,这里的高血铁蛋白症与高转铁蛋白饱和度、铁储存巨噬细胞和进行性实质细胞铁负荷有关。腹部磁共振成像(MRI)是 FD 诊断的关键非侵入性诊断工具,显示特征性的铁负荷 SSL 三联征(脾脏、脊柱和肝脏)。建议采用非侵袭性的放血方案,并由于贫血的风险,需要仔细监测转铁蛋白饱和度和血红蛋白。由于兄弟姐妹和后代有 50%的机会携带致病性突变,因此必须进行家族筛查。