1Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD, USA.
2Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, USA.
Microb Genom. 2017 Jul 8;3(9):e000122. doi: 10.1099/mgen.0.000122. eCollection 2017 Sep.
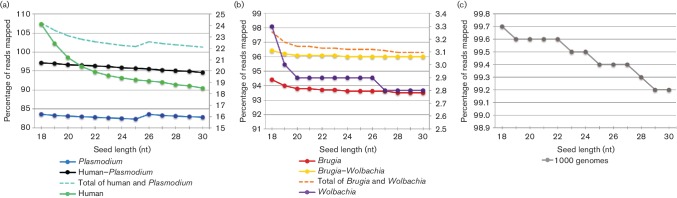
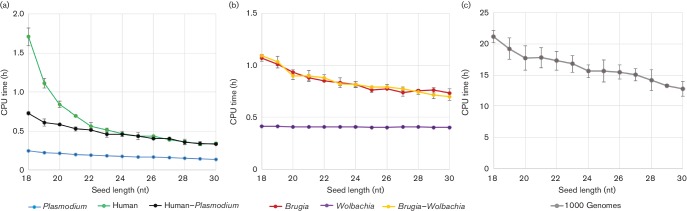
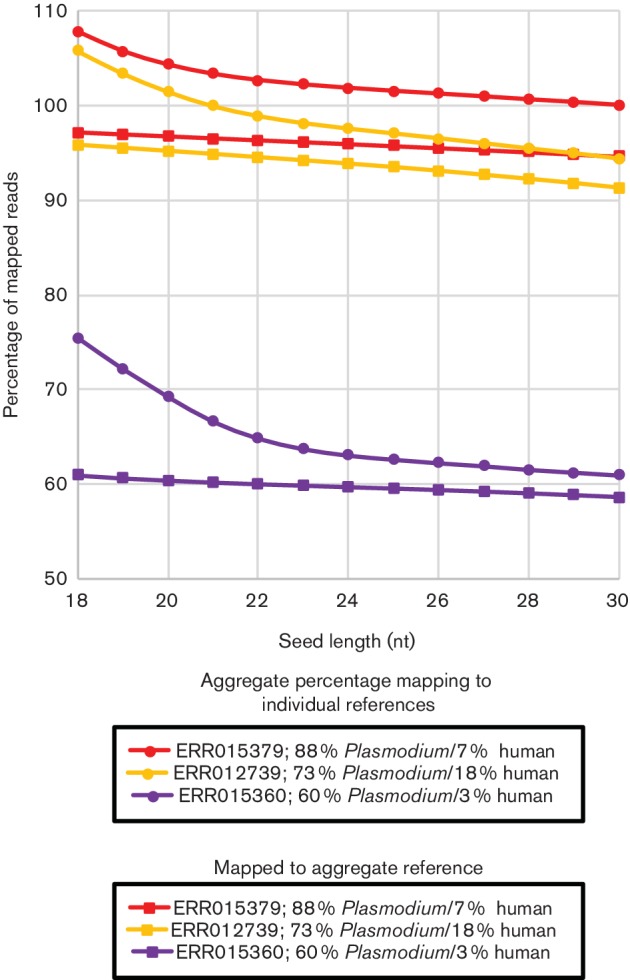
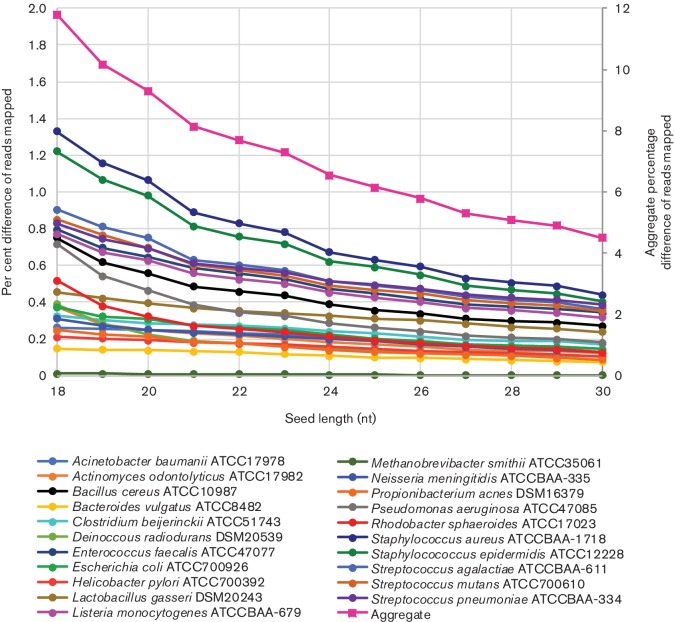
As sequencing technologies have evolved, the tools to analyze these sequences have made similar advances. However, for multi-species samples, we observed important and adverse differences in alignment specificity and computation time for bwa- mem (Burrows-Wheeler aligner-maximum exact matches) relative to bwa-aln. Therefore, we sought to optimize bwa-mem for alignment of data from multi-species samples in order to reduce alignment time and increase the specificity of alignments. In the multi-species cases examined, there was one majority member (i.e. or ) and one minority member (i.e. human or the endosymbiont Bm) of the sequence data. Increasing bwa-mem seed length from the default value reduced the number of read pairs from the majority sequence member that incorrectly aligned to the reference genome of the minority sequence member. Combining both source genomes into a single reference genome increased the specificity of mapping, while also reducing the central processing unit (CPU) time. In , at a seed length of 18 nt, 24.1 % of reads mapped to the human genome using 1.7±0.1 CPU hours, while 83.6 % of reads mapped to the genome using 0.2±0.0 CPU hours (total: 107.7 % reads mapping; in 1.9±0.1 CPU hours). In contrast, 97.1 % of the reads mapped to a combined human reference in only 0.7±0.0 CPU hours. Overall, the results suggest that combining all references into a single reference database and using a 23 nt seed length reduces the computational time, while maximizing specificity. Similar results were found for simulated sequence reads from a mock metagenomic data set. We found similar improvements to computation time in a publicly available human-only data set.
随着测序技术的发展,分析这些序列的工具也取得了类似的进展。然而,对于多物种样本,我们观察到 bwa-mem(Burrows-Wheeler aligner-maximum exact matches)相对于 bwa-aln 在对齐特异性和计算时间方面存在重要且不利的差异。因此,我们试图优化 bwa-mem 以对齐多物种样本的数据,以减少对齐时间并提高对齐的特异性。在所检查的多物种情况下,序列数据有一个主要成员(即 或 )和一个少数成员(即人类或内共生体 Bm)。将 bwa-mem 的种子长度从默认值增加,可以减少来自主要序列成员的读对数量,这些读对错误地与少数序列成员的参考基因组对齐。将两个源基因组合并到一个单一的参考基因组中,提高了映射的特异性,同时也减少了中央处理器(CPU)时间。在 中,在种子长度为 18nt 的情况下,使用 1.7±0.1 CPU 小时,有 24.1%的读对映射到人类基因组,而使用 0.2±0.0 CPU 小时,有 83.6%的读对映射到 基因组(总共:107.7%的读对映射;在 1.9±0.1 CPU 小时内)。相比之下,在仅使用 0.7±0.0 CPU 小时的情况下,97.1%的读对映射到一个组合的人类参考。总体而言,结果表明,将所有参考合并到一个单一的参考数据库中,并使用 23nt 的种子长度可以减少计算时间,同时最大限度地提高特异性。在模拟的宏基因组数据集的序列读取中也发现了类似的结果。我们在一个公开的仅人类数据集发现了计算时间的类似改进。