Falco Mariateresa, Amabile Sonia, Acquaviva Fabio
Department of Molecular Medicine and Medical Biotechnology, University of Naples "Federico II", Naples, Italy.
Department of Translational Medical Sciences (DISMET), Section of Pediatric Clinical Genetics, University of Naples "Federico II", Naples, Italy.
Appl Clin Genet. 2017 Nov 3;10:85-94. doi: 10.2147/TACG.S128455. eCollection 2017.
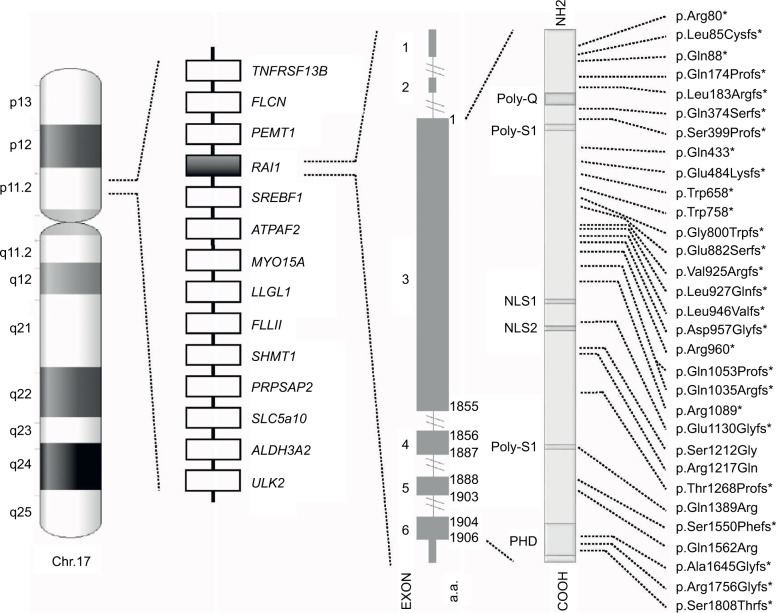
Smith-Magenis syndrome (SMS; OMIM #182290) is a complex genetic disorder characterized by distinctive physical features, developmental delay, cognitive impairment, and a typical behavioral phenotype. SMS is caused by interstitial 17p11.2 deletions, encompassing multiple genes and including the retinoic acid-induced 1 gene (), or by mutations in itself. About 10% of all the SMS patients, in fact, carry an mutation responsible for the phenotype. (OMIM *607642) is a dosage-sensitive gene expressed in many tissues and highly conserved among species. Over the years, several studies have demonstrated that (or its homologs in animal models) acts as a transcriptional factor implicated in embryonic neurodevelopment, neuronal differentiation, cell growth and cell cycle regulation, bone and skeletal development, lipid and glucose metabolisms, behavioral functions, and circadian activity. Patients with pathogenic variants show some phenotypic differences when compared to those carrying the typical deletion. They usually have lower incidence of hypotonia and less cognitive impairment than those with 17p11.2 deletions but more frequently show the behavioral characteristics of the syndrome and overeating issues. These differences reflect the primary pathogenetic role of without the pathogenetic contribution of the other genes included in the typical 17p11.2 deletion. The better comprehension of physiological roles of , its molecular co-workers and interactors, and its contribution in determining the typical SMS phenotype will certainly open a new path for therapeutic interventions.
史密斯-马吉尼斯综合征(SMS;OMIM #182290)是一种复杂的遗传性疾病,其特征为独特的身体特征、发育迟缓、认知障碍和典型的行为表型。SMS由17号染色体短臂11.2区间质缺失引起,该缺失包含多个基因,包括视黄酸诱导1基因(),或由自身的突变引起。事实上,所有SMS患者中约10%携带导致该表型的突变。(OMIM *607642)是一个剂量敏感基因,在许多组织中表达,且在物种间高度保守。多年来,多项研究表明(或其在动物模型中的同源物)作为一种转录因子,参与胚胎神经发育、神经元分化、细胞生长和细胞周期调控、骨骼发育、脂质和葡萄糖代谢、行为功能以及昼夜节律活动。与携带典型缺失的患者相比,携带致病变异的患者表现出一些表型差异。他们通常比17p11.2缺失的患者肌张力低下的发生率更低,认知障碍也更少,但更频繁地表现出该综合征的行为特征和暴饮暴食问题。这些差异反映了在没有典型17p11.2缺失中其他基因致病作用情况下的主要致病作用。更好地理解的生理作用、其分子协同作用因子和相互作用分子,以及其在确定典型SMS表型中的作用,必将为治疗干预开辟一条新途径。