Petzold Markus, Prior Karola, Moran-Gilad Jacob, Harmsen Dag, Lück Christian
Institute of Medical Microbiology and Hygiene, Dresden University of Technology, Dresden, Germany.
These authors contributed equally to the work.
Euro Surveill. 2017 Nov;22(45). doi: 10.2807/1560-7917.ES.2017.22.45.17-00137.
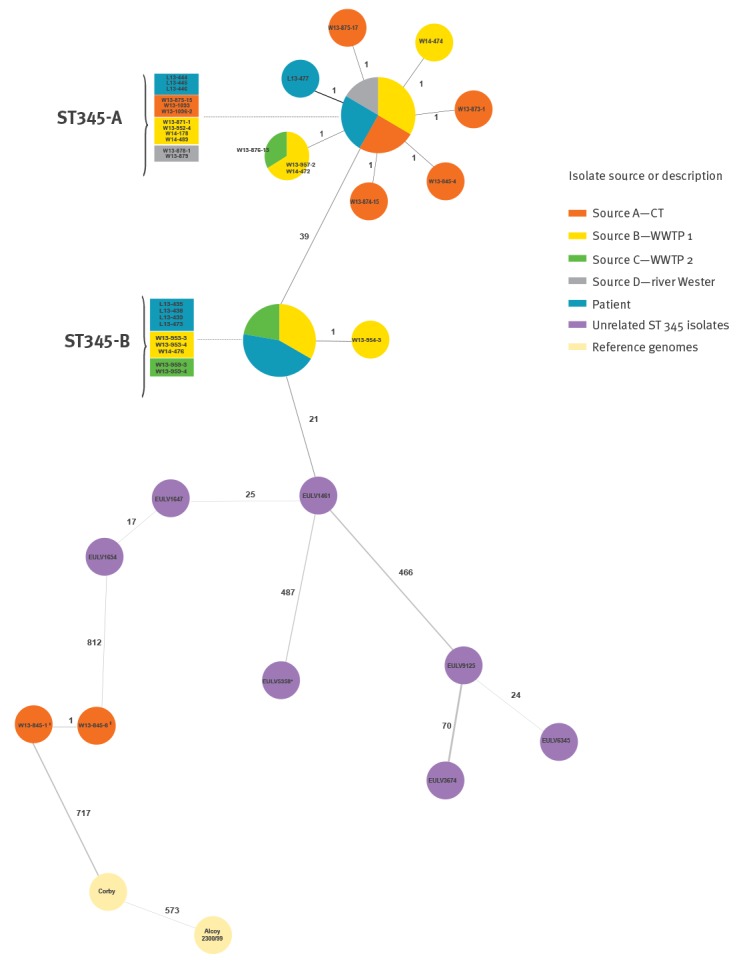
IntroductionWhole genome sequencing (WGS) is increasingly used in Legionnaires' disease (LD) outbreak investigations, owing to its higher resolution than sequence-based typing, the gold standard typing method for in the analysis of endemic strains. Recently, a gene-by-gene typing approach based on 1,521 core genes called core genome multilocus sequence typing (cgMLST) was described that enables a robust and standardised typing of . : We applied this cgMLST scheme to isolates obtained during the largest outbreak of LD reported so far in Germany. In this outbreak, the epidemic clone ST345 had been isolated from patients and four different environmental sources. In total 42 clinical and environmental isolates were retrospectively typed. : Epidemiologically unrelated ST345 isolates were clearly distinguishable from the epidemic clone. Remarkably, epidemic isolates split up into two distinct clusters, ST345-A and ST345-B, each respectively containing a mix of clinical and epidemiologically-related environmental samples. : The outbreak was therefore likely caused by both variants of the single sequence type, which pre-existed in the environmental reservoirs. The two clusters differed by 40 alleles located in two neighbouring genomic regions of ca 42 and 26 kb. Additional analysis supported horizontal gene transfer of the two regions as responsible for the difference between the variants. Both regions comprise virulence genes and have previously been reported to be involved in recombination events. This corroborates the notion that genomic outbreak investigations should always take epidemiological information into consideration when making inferences. Overall, cgMLST proved helpful in disentangling the complex genomic epidemiology of the outbreak.
引言
全基因组测序(WGS)在军团病(LD)暴发调查中的应用越来越多,这是因为其分辨率高于基于序列的分型方法,而基于序列的分型是分析地方性菌株的金标准分型方法。最近,一种基于1521个核心基因的逐个基因分型方法——核心基因组多位点序列分型(cgMLST)被提出,该方法能够对……进行可靠且标准化的分型。我们将这种cgMLST方案应用于德国迄今报告的最大规模LD暴发期间分离得到的菌株。在此次暴发中,流行克隆ST345已从患者和四个不同环境来源中分离出来。总共对42株临床和环境分离株进行了回顾性分型。在流行病学上不相关的ST345分离株与流行克隆明显可区分。值得注意的是,流行分离株分为两个不同的簇,ST345 - A和ST345 - B,每个簇分别包含临床和与流行病学相关的环境样本的混合。因此,此次暴发可能是由单一序列型的两种变体引起的,这两种变体预先存在于环境储库中。这两个簇在位于约42 kb和26 kb的两个相邻基因组区域中的40个等位基因上存在差异。进一步分析支持这两个区域的水平基因转移是导致变体间差异的原因。这两个区域都包含毒力基因,并且先前已报道它们参与重组事件。这证实了这样一种观点,即在进行推断时,基因组暴发调查应始终考虑流行病学信息。总体而言,cgMLST被证明有助于理清此次暴发复杂的基因组流行病学情况。