Nagata Shohei, Imai Junnosuke, Makino Gakuto, Tomita Masaru, Kanai Akio
Institute for Advanced Biosciences, Keio University, Tsuruoka, Japan.
Faculty of Environment and Information Studies, Keio University, Fujisawa, Japan.
Front Microbiol. 2017 Nov 2;8:2151. doi: 10.3389/fmicb.2017.02151. eCollection 2017.
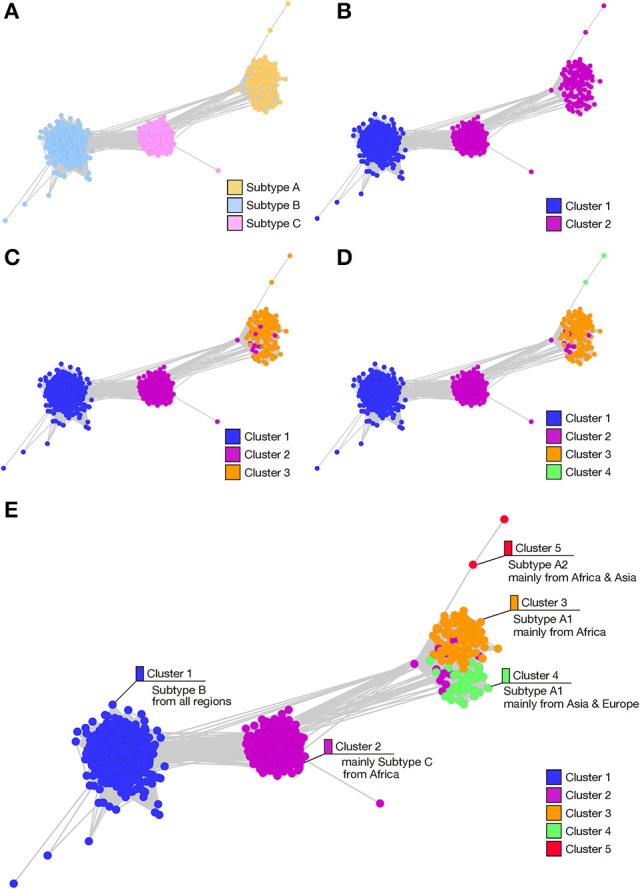
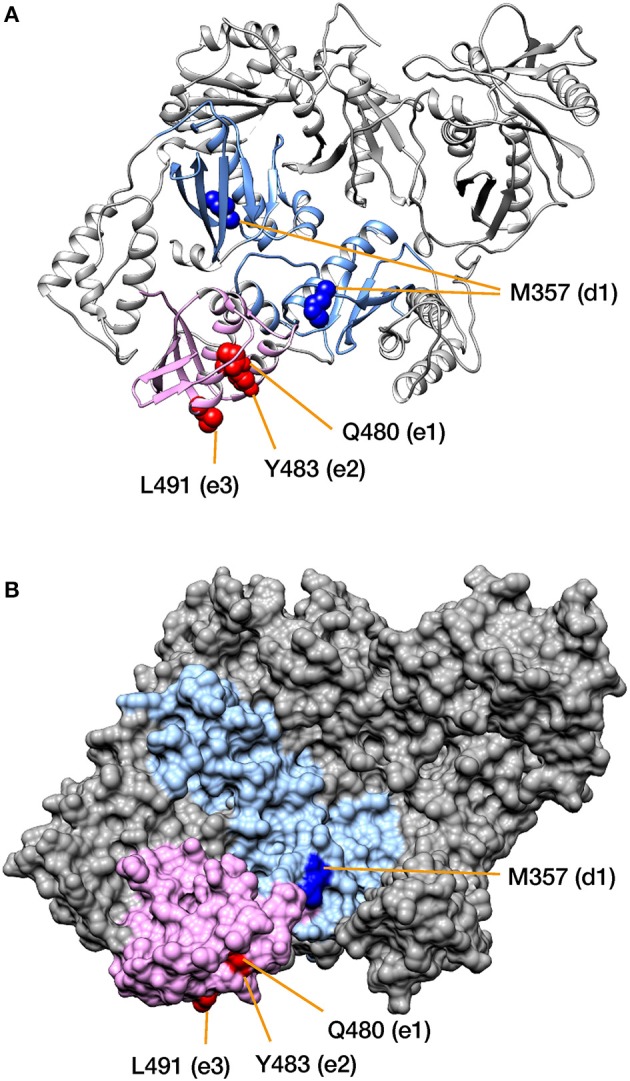
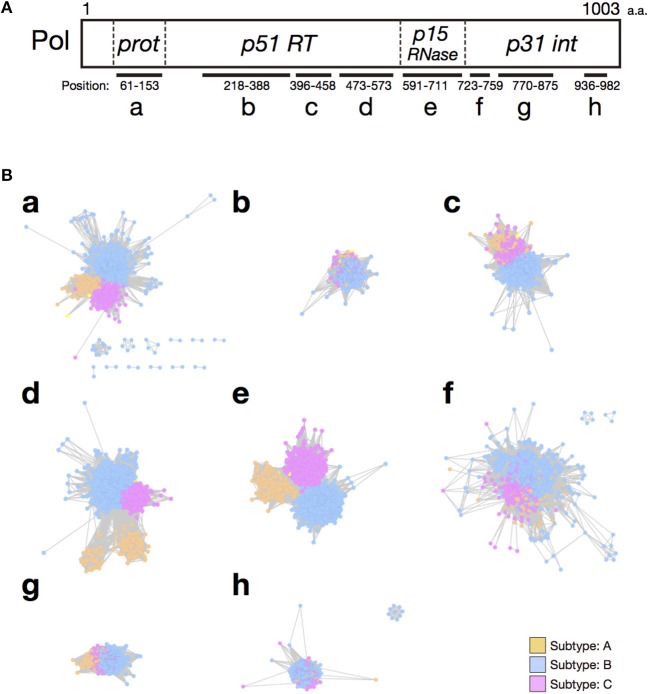
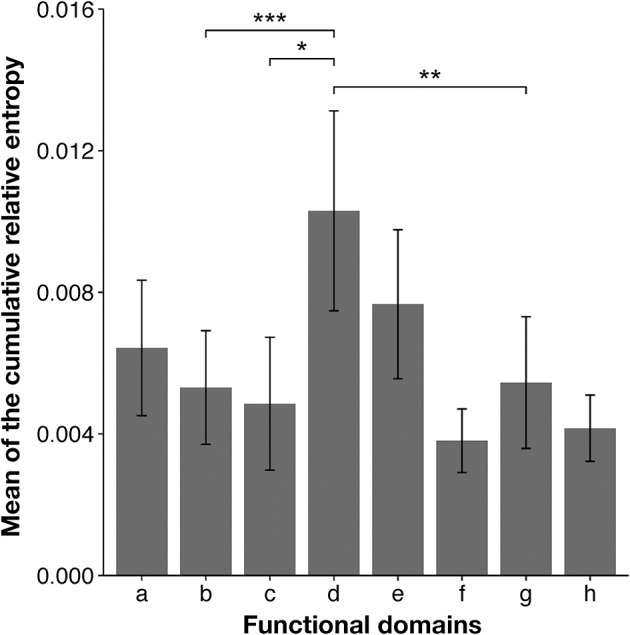
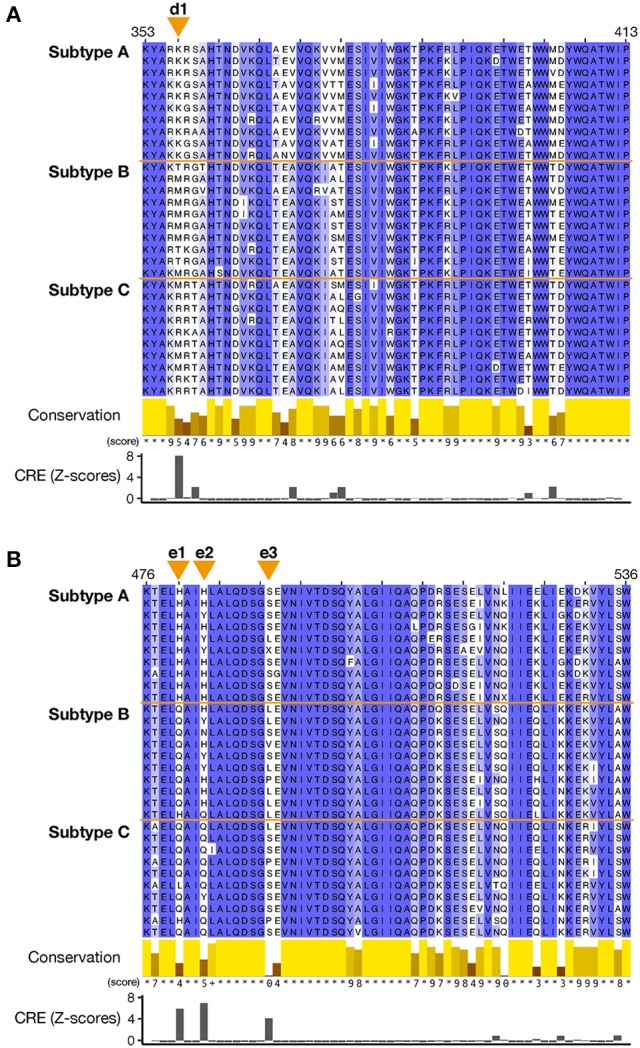
RNA viruses have been used as model systems to understand the patterns and processes of molecular evolution because they have high mutation rates and are genetically diverse. (HIV-1), the etiological agent of acquired immune deficiency syndrome, is highly genetically diverse, and is classified into several groups and subtypes. However, it has been difficult to use its diverse sequences to establish the overall phylogenetic relationships of different strains or the trends in sequence conservation with the construction of phylogenetic trees. Our aims were to systematically characterize HIV-1 subtype evolution and to identify the regions responsible for HIV-1 subtype differentiation at the amino acid level in the Pol protein, which is often used to classify the HIV-1 subtypes. In this study, we systematically characterized the mutation sites in 2,052 Pol proteins from HIV-1 group M (144 subtype A; 1,528 subtype B; 380 subtype C), using sequence similarity networks. We also used spectral clustering to group the sequences based on the network graph structures. A stepwise analysis of the cluster hierarchies allowed us to estimate a possible evolutionary pathway for the Pol proteins. The subtype A sequences also clustered according to when and where the viruses were isolated, whereas both the subtype B and C sequences remained as single clusters. Because the Pol protein has several functional domains, we identified the regions that are discriminative by comparing the structures of the domain-based networks. Our results suggest that sequence changes in the RNase H domain and the reverse transcriptase (RT) connection domain are responsible for the subtype classification. By analyzing the different amino acid compositions at each site in both domain sequences, we found that a few specific amino acid residues (i.e., M357 in the RT connection domain and Q480, Y483, and L491 in the RNase H domain) represent the differences among the subtypes. These residues were located on the surface of the RT structure and in the vicinity of the amino acid sites responsible for RT enzymatic activity or function.
RNA病毒已被用作模型系统来理解分子进化的模式和过程,因为它们具有高突变率且基因多样。获得性免疫缺陷综合征的病原体人类免疫缺陷病毒1型(HIV-1)基因高度多样,可分为几个组和亚型。然而,利用其多样的序列通过构建系统发育树来建立不同毒株的整体系统发育关系或序列保守性趋势一直很困难。我们的目标是系统地表征HIV-1亚型的进化,并在常用于HIV-1亚型分类的Pol蛋白中,在氨基酸水平上鉴定负责HIV-1亚型分化的区域。在本研究中,我们使用序列相似性网络系统地表征了来自HIV-1 M组的2052个Pol蛋白中的突变位点(144个A亚型;1528个B亚型;380个C亚型)。我们还使用光谱聚类根据网络图结构对序列进行分组。对聚类层次的逐步分析使我们能够估计Pol蛋白可能的进化途径。A亚型序列也根据病毒分离的时间和地点聚类,而B亚型和C亚型序列仍为单个聚类。由于Pol蛋白有几个功能域,我们通过比较基于结构域的网络结构来鉴定具有区分性的区域。我们的结果表明,核糖核酸酶H结构域和逆转录酶(RT)连接结构域中的序列变化负责亚型分类。通过分析两个结构域序列中每个位点的不同氨基酸组成,我们发现一些特定的氨基酸残基(即RT连接结构域中的M357以及核糖核酸酶H结构域中的Q480、Y483和L491)代表了亚型之间的差异。这些残基位于RT结构的表面以及负责RT酶活性或功能的氨基酸位点附近。