Department of Basic and Clinical Neuroscience, King's College London, 125 Coldharbour Lane, SE5 9NU London, UK.
Department of Clinical Genetics and Alzheimer Center, VU University Medical Center, Amsterdam, the Netherlands; Department of Functional Genome Analysis, VU University, Amsterdam, the Netherlands.
Curr Biol. 2017 Dec 4;27(23):3626-3642.e6. doi: 10.1016/j.cub.2017.10.054. Epub 2017 Nov 22.
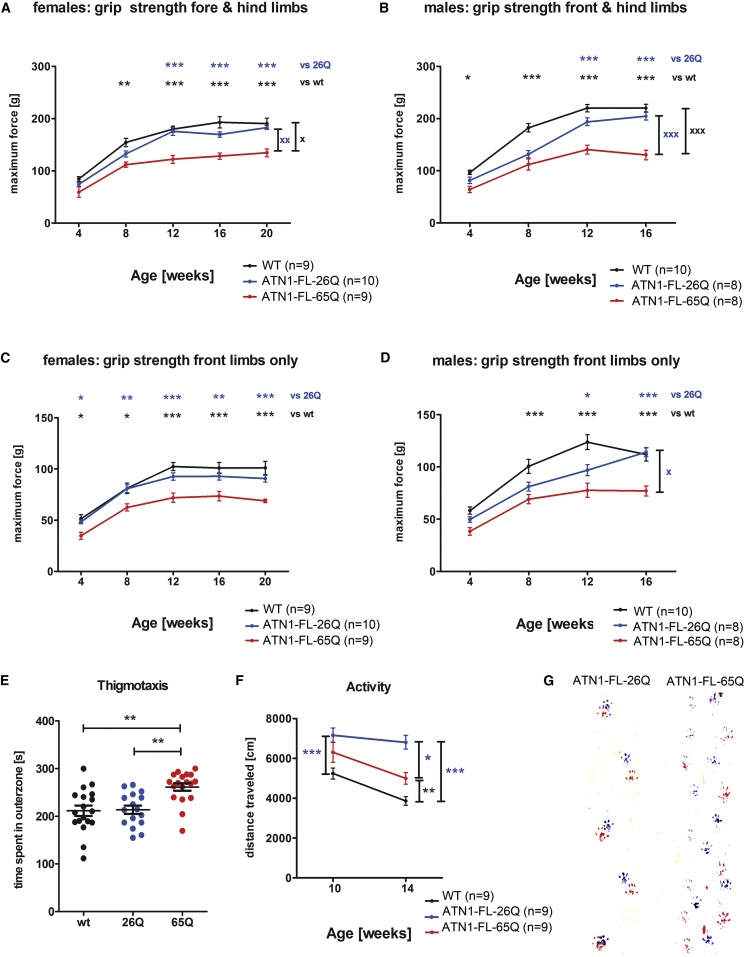
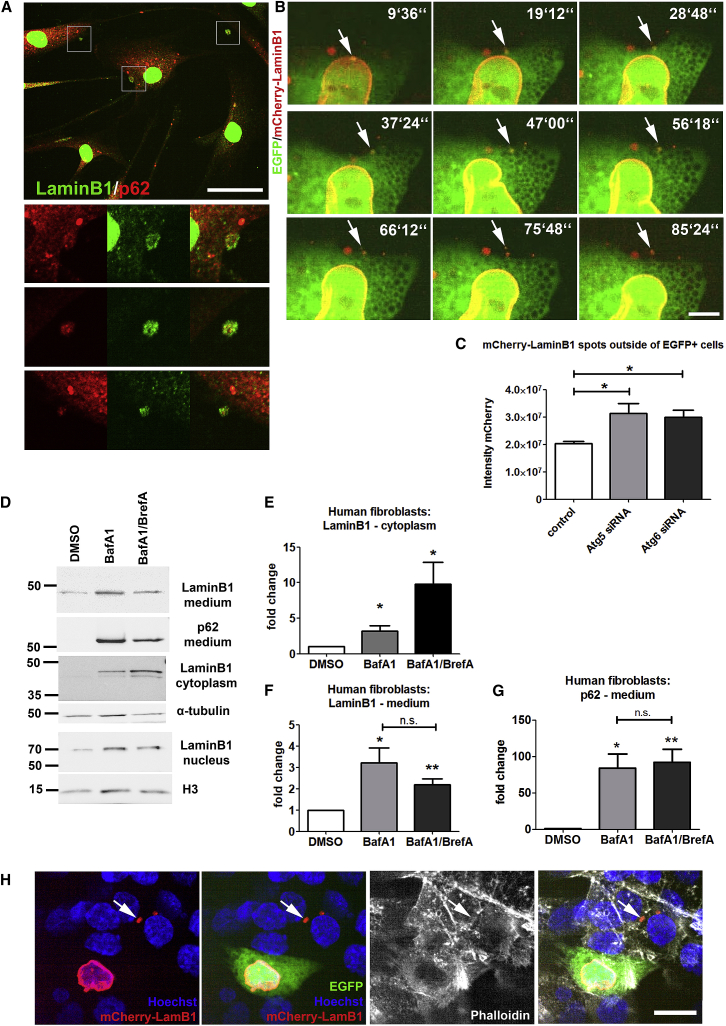
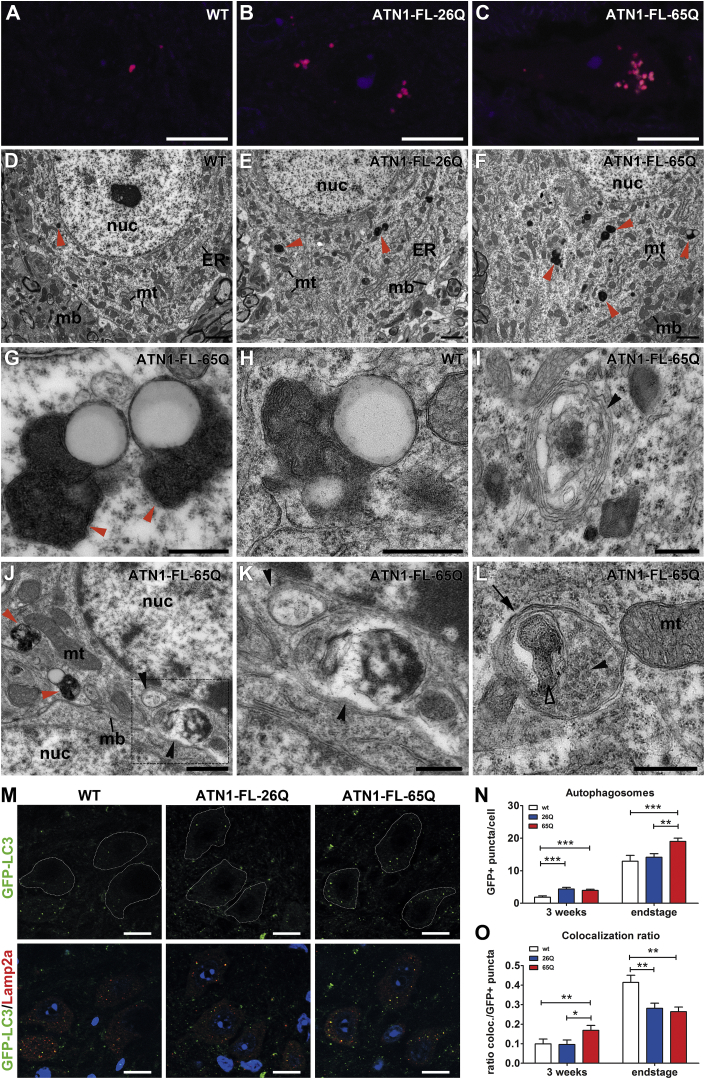
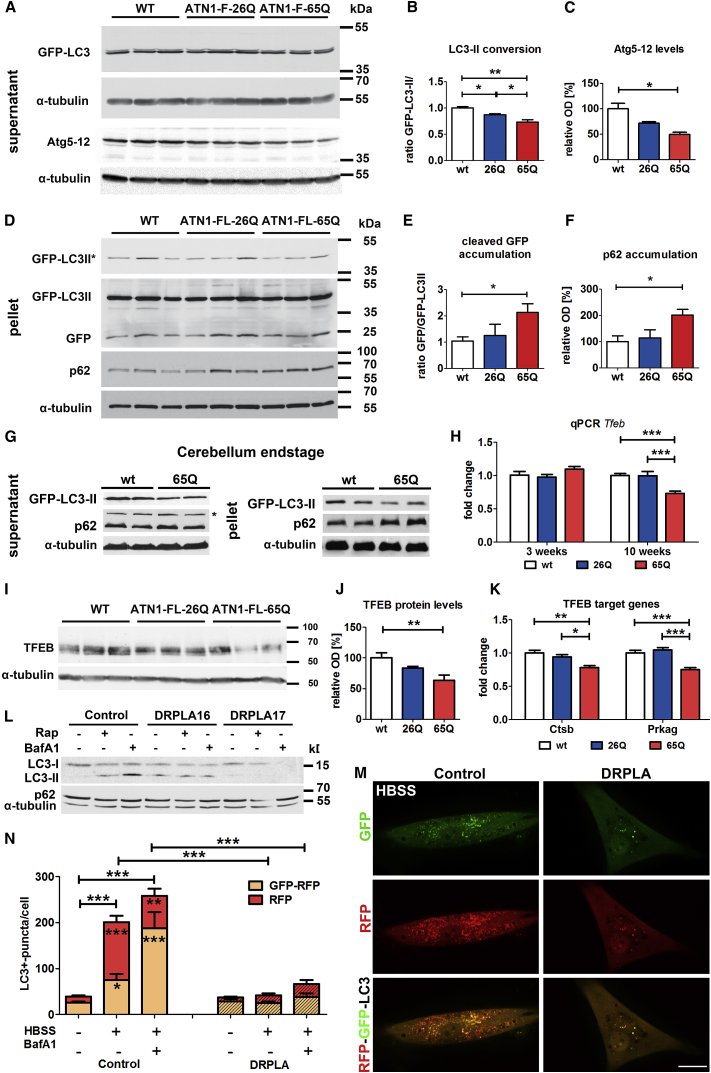
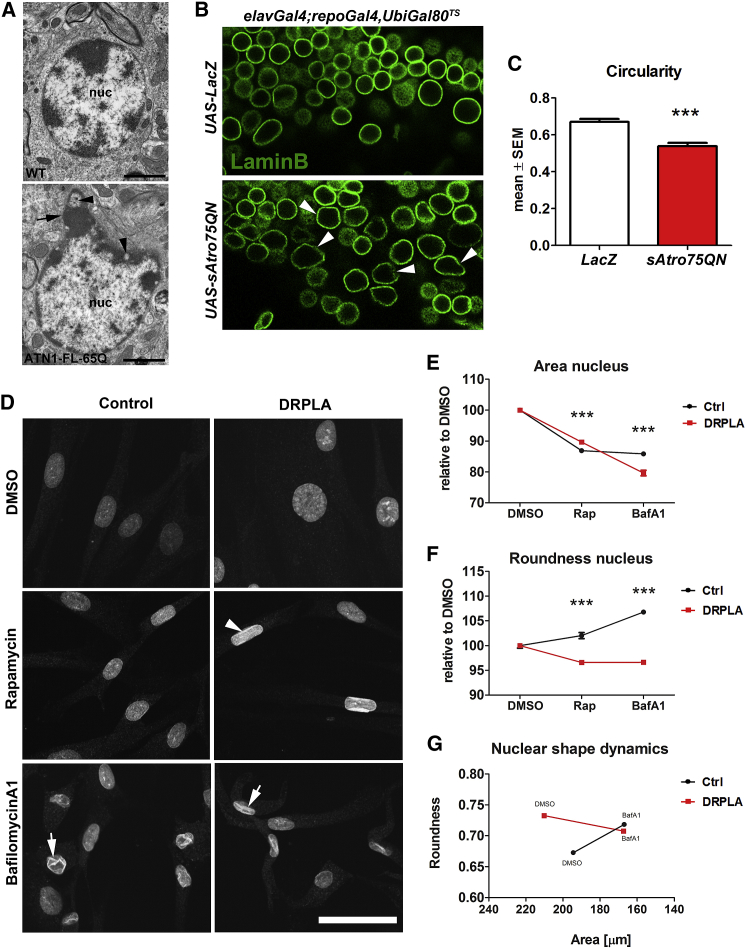
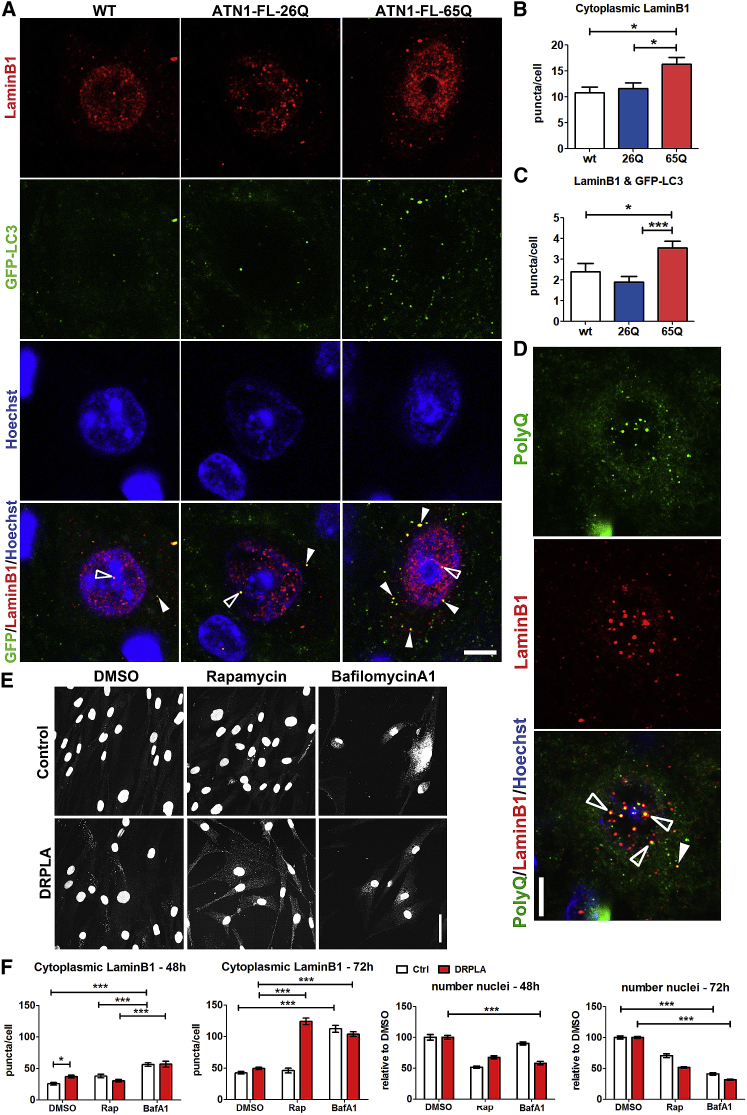
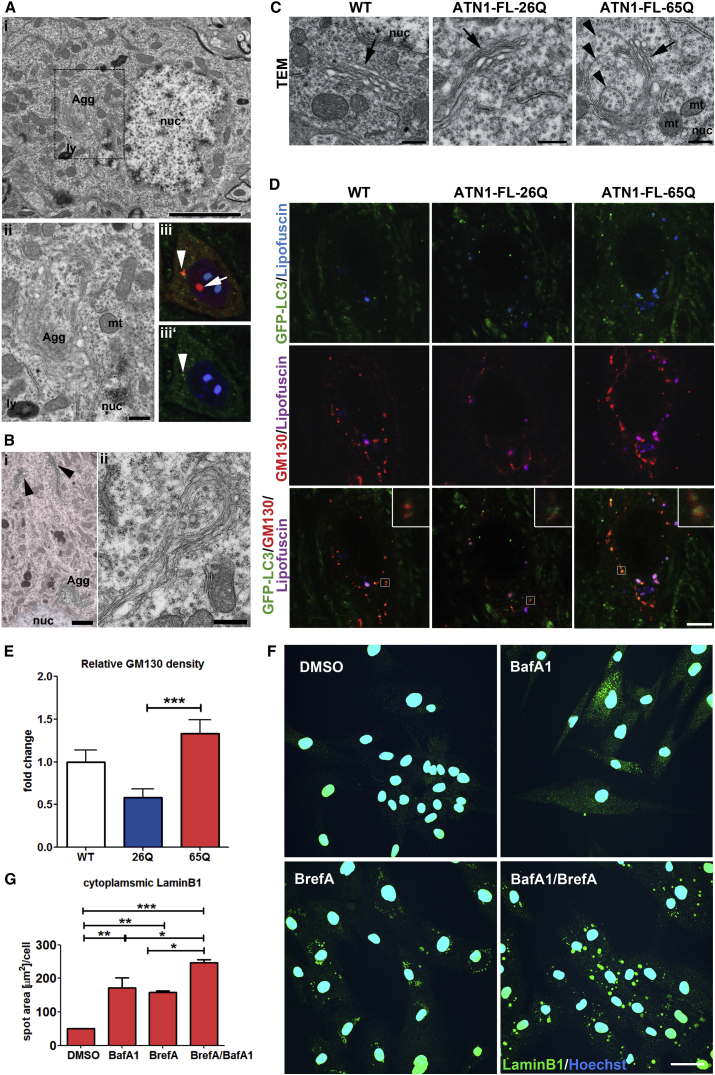
The terminal stages of neuronal degeneration and death in neurodegenerative diseases remain elusive. Autophagy is an essential catabolic process frequently failing in neurodegeneration. Selective autophagy routes have recently emerged, including nucleophagy, defined as degradation of nuclear components by autophagy. Here, we show that, in a mouse model for the polyglutamine disease dentatorubral-pallidoluysian atrophy (DRPLA), progressive acquirement of an ataxic phenotype is linked to severe cerebellar cellular pathology, characterized by nuclear degeneration through nucleophagy-based LaminB1 degradation and excretion. We find that canonical autophagy is stalled in DRPLA mice and in human fibroblasts from patients of DRPLA. This is evidenced by accumulation of p62 and downregulation of LC3-I/II conversion as well as reduced Tfeb expression. Chronic autophagy blockage in several conditions, including DRPLA and Vici syndrome, an early-onset autolysosomal pathology, leads to the activation of alternative clearance pathways including Golgi membrane-associated and nucleophagy-based LaminB1 degradation and excretion. The combination of these alternative pathways and canonical autophagy blockade, results in dramatic nuclear pathology with disruption of the nuclear organization, bringing about terminal cell atrophy and degeneration. Thus, our findings identify a novel progressive mechanism for the terminal phases of neuronal cell degeneration and death in human neurodegenerative diseases and provide a link between autophagy block, activation of alternative pathways for degradation, and excretion of cellular components.
神经退行性疾病中神经元退化和死亡的终末阶段仍然难以捉摸。自噬是一种重要的分解代谢过程,在神经退行性变中经常失败。最近出现了选择性自噬途径,包括核噬作用,定义为通过自噬降解核成分。在这里,我们表明,在多聚谷氨酰胺疾病齿状核红核苍白球路易体萎缩症(DRPLA)的小鼠模型中,进行性获得共济失调表型与严重的小脑细胞病理学有关,其特征是通过核噬作用的核纤层蛋白 B1 降解和排泄导致核退化。我们发现,在 DRPLA 小鼠和 DRPLA 患者的成纤维细胞中,经典自噬被停滞。这可以通过 p62 的积累和 LC3-I/II 转化的下调以及 Tfeb 表达的减少来证明。几种情况下的慢性自噬阻断,包括 DRPLA 和 Vici 综合征,一种早期的自噬溶酶体病理学,导致替代清除途径的激活,包括高尔基膜相关和核噬作用的核纤层蛋白 B1 降解和排泄。这些替代途径和经典自噬阻断的结合导致明显的核病理学,破坏核组织,导致终末细胞萎缩和退化。因此,我们的发现确定了人类神经退行性疾病中神经元细胞退化和死亡的终末阶段的一种新的进行性机制,并提供了自噬阻断、替代降解途径的激活以及细胞成分的排泄之间的联系。