Sato Mitsuharu, Miyazaki Kentaro
Bioproduction Research Institute, Department of Life Science and Biotechnology, National Institute of Advanced Industrial Science and Technology, Tsukuba, Japan.
Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Japan.
Front Microbiol. 2017 Nov 16;8:2225. doi: 10.3389/fmicb.2017.02225. eCollection 2017.
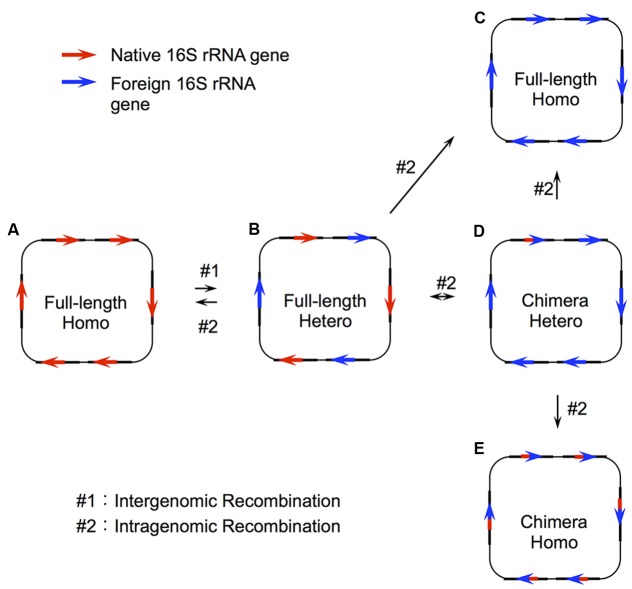
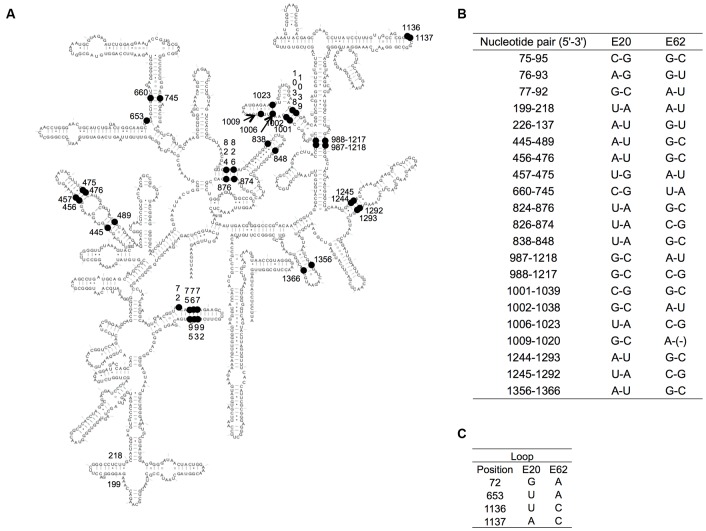
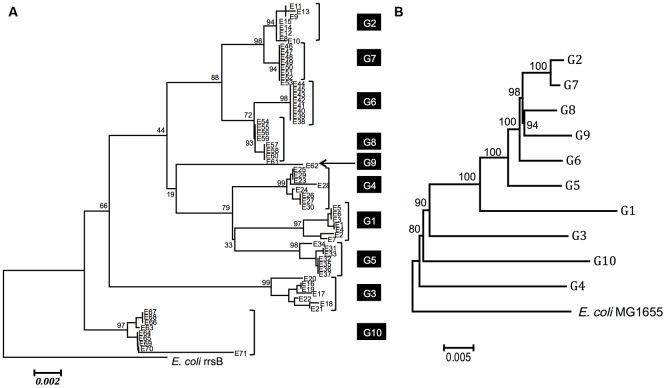
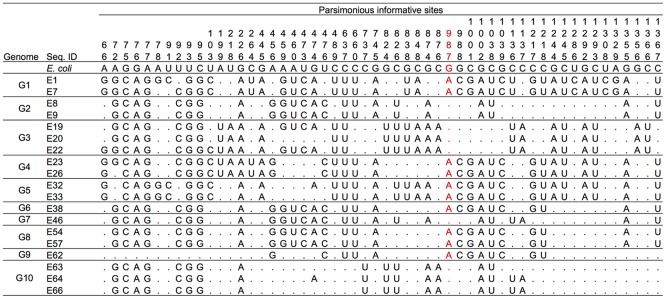
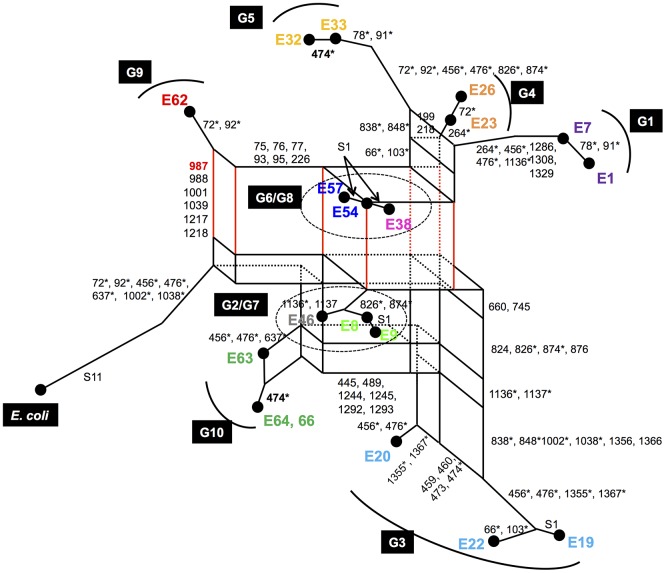
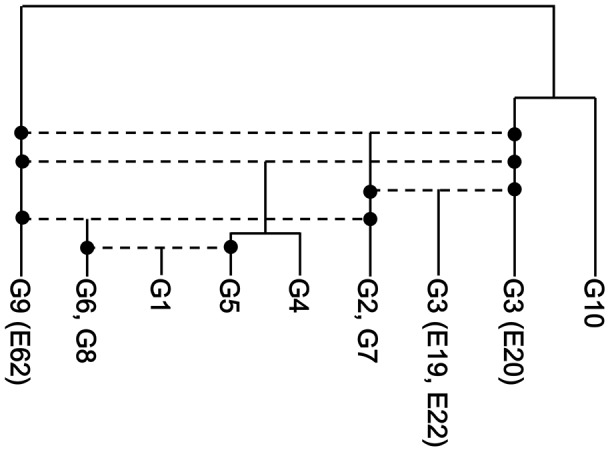
Horizontal gene transfer (HGT) is a ubiquitous genetic event in bacterial evolution, but it seldom occurs for genes involved in highly complex supramolecules (or biosystems), which consist of many gene products. The ribosome is one such supramolecule, but several bacteria harbor dissimilar and/or chimeric 16S rRNAs in their genomes, suggesting the occurrence of HGT of this gene. However, we know little about whether the genes actually experience HGT and, if so, the frequency of such a transfer. This is primarily because the methods currently employed for phylogenetic analysis (e.g., neighbor-joining, maximum likelihood, and maximum parsimony) of 16S rRNA genes assume point mutation-driven tree-shape evolution as an evolutionary model, which is intrinsically inappropriate to decipher the evolutionary history for genes driven by recombination. To address this issue, we applied a phylogenetic network analysis, which has been used previously for detection of genetic recombination in homologous alleles, to the 16S rRNA gene. We focused on the genus , whose phylogenetic relationships inferred by multi-locus sequence alignment analysis and 16S rRNA sequences are incompatible. All 10 complete genomic sequences were retrieved from the NCBI database, in which 71 16S rRNA genes were included. Neighbor-joining analysis demonstrated that the genes residing in the same genomes clustered, indicating the occurrence of intragenomic recombination. However, as suggested by the low bootstrap values, evolutionary relationships between the clusters were uncertain. We then applied phylogenetic network analysis to representative sequences from each cluster. We found three ancestral 16S rRNA groups; the others were likely created through recursive recombination between the ancestors and chimeric descendants. Despite the large sequence changes caused by the recombination events, the RNA secondary structures were conserved. Successive intergenomic and intragenomic recombination thus shaped the evolution of 16S rRNA genes in the genus .
水平基因转移(HGT)是细菌进化中普遍存在的遗传事件,但对于涉及由许多基因产物组成的高度复杂超分子(或生物系统)的基因来说,这种情况很少发生。核糖体就是这样一种超分子,但几种细菌的基因组中存在不同和/或嵌合的16S rRNA,这表明该基因发生了HGT。然而,我们对于这些基因是否真的经历了HGT以及如果是这样,这种转移的频率了解甚少。这主要是因为目前用于16S rRNA基因系统发育分析(例如邻接法、最大似然法和最大简约法)的方法将点突变驱动的树形进化作为进化模型,而这种模型本质上不适用于解读由重组驱动的基因的进化历史。为了解决这个问题,我们将先前用于检测同源等位基因中基因重组的系统发育网络分析应用于16S rRNA基因。我们聚焦于一个属,其通过多位点序列比对分析和16S rRNA序列推断出的系统发育关系并不一致。从NCBI数据库中检索到了所有10个完整基因组序列,其中包含71个16S rRNA基因。邻接法分析表明,位于相同基因组中的基因聚集在一起,表明发生了基因组内重组。然而,正如低自展值所表明的那样,各聚类之间的进化关系并不确定。然后我们将系统发育网络分析应用于每个聚类的代表性序列。我们发现了三个祖先16S rRNA组;其他的可能是通过祖先和嵌合后代之间的递归重组产生的。尽管重组事件导致了大量的序列变化,但RNA二级结构是保守的。因此,连续的基因组间和基因组内重组塑造了该属中16S rRNA基因的进化。