Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Division of Genetics and Genomics and Howard Hughes Medical Institute, Boston Children's Hospital, Boston, MA 02115, USA; Departments of Neurology and Pediatrics, Harvard Medical School, Boston, MA 02115, USA; Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Nucleic Acids Res. 2018 Feb 28;46(4):e20. doi: 10.1093/nar/gkx1195.
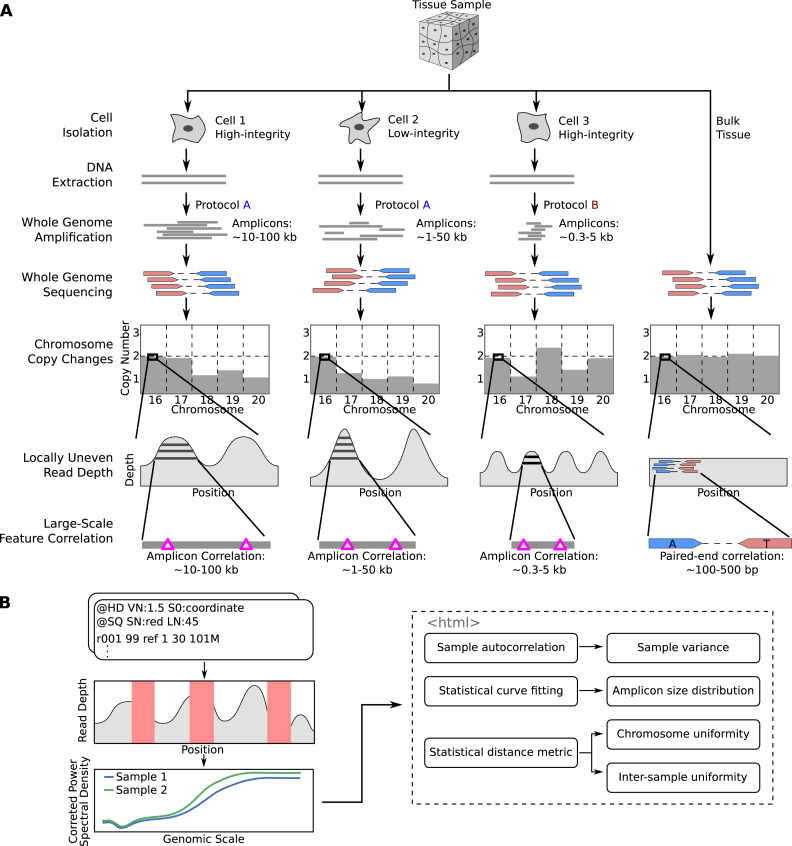
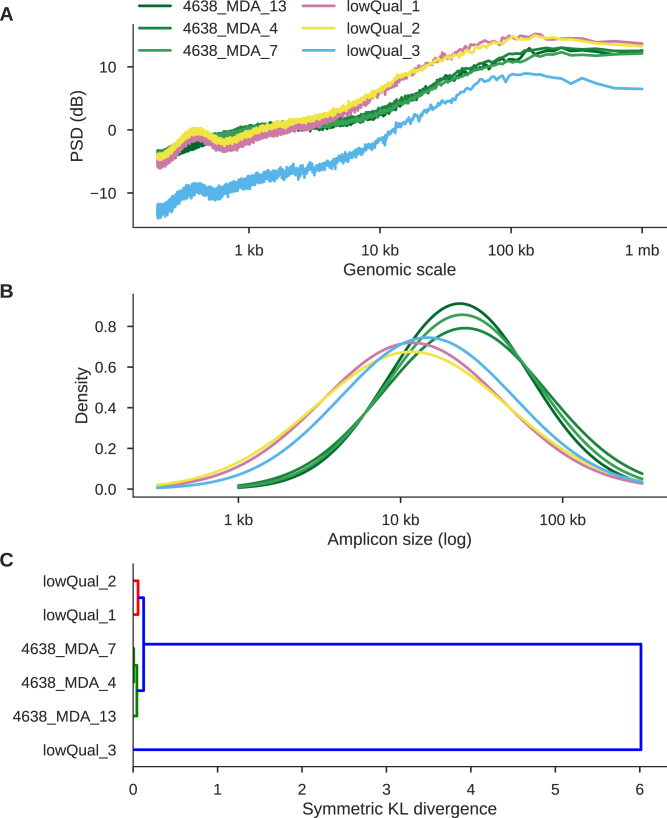
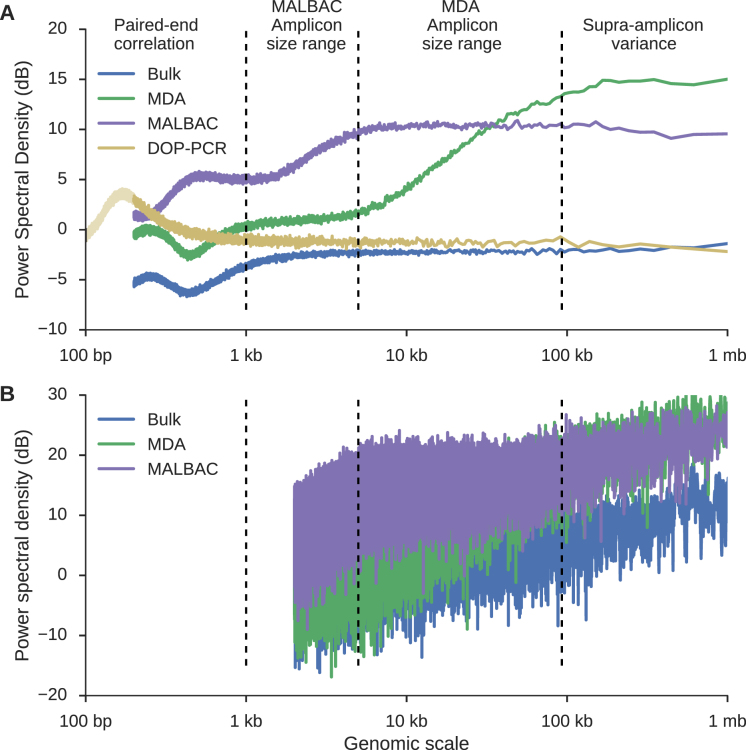
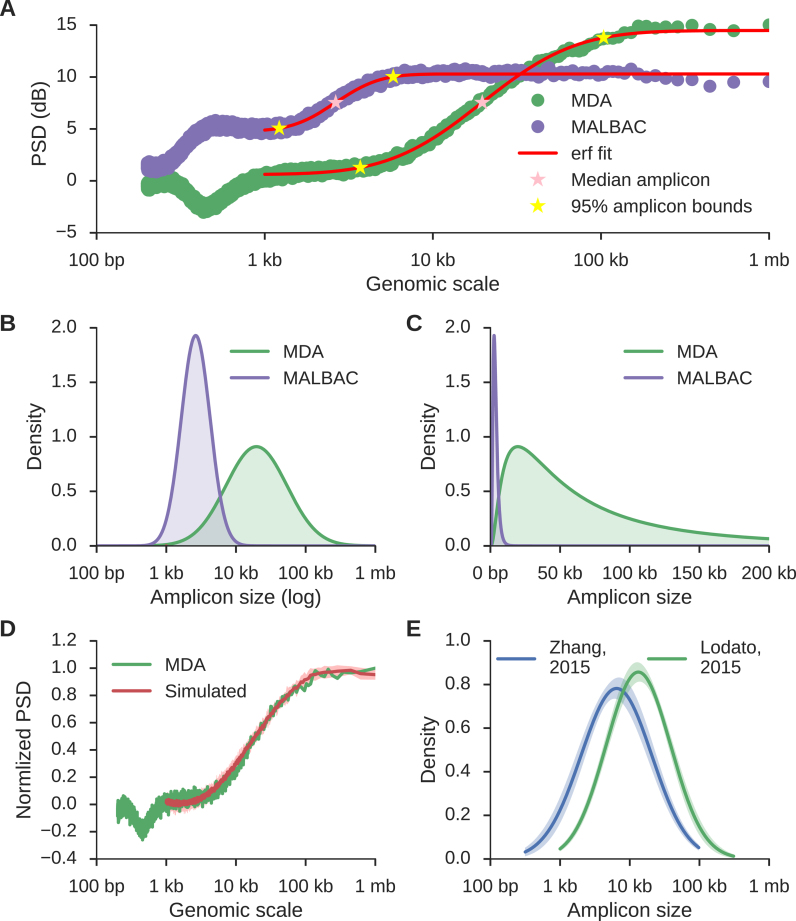
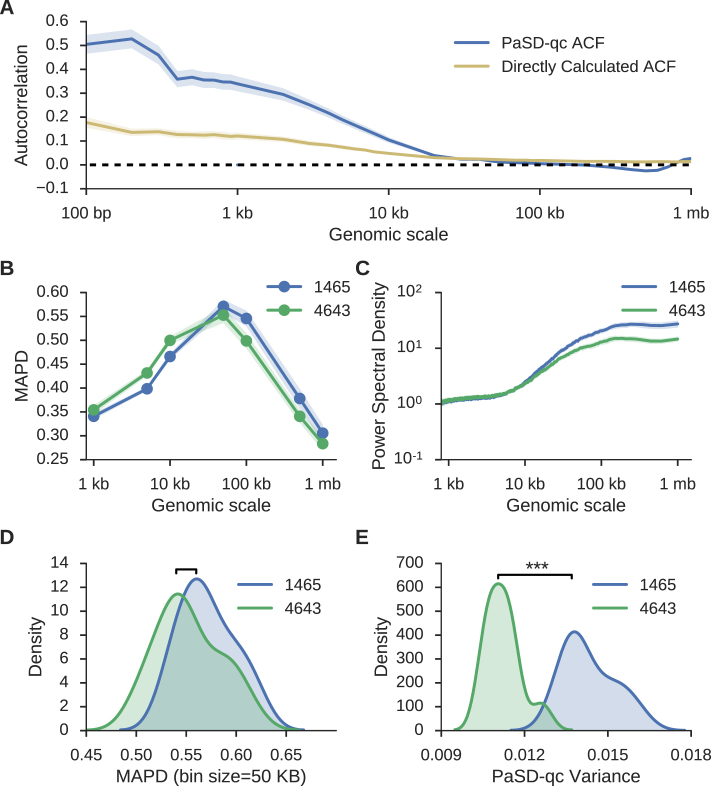
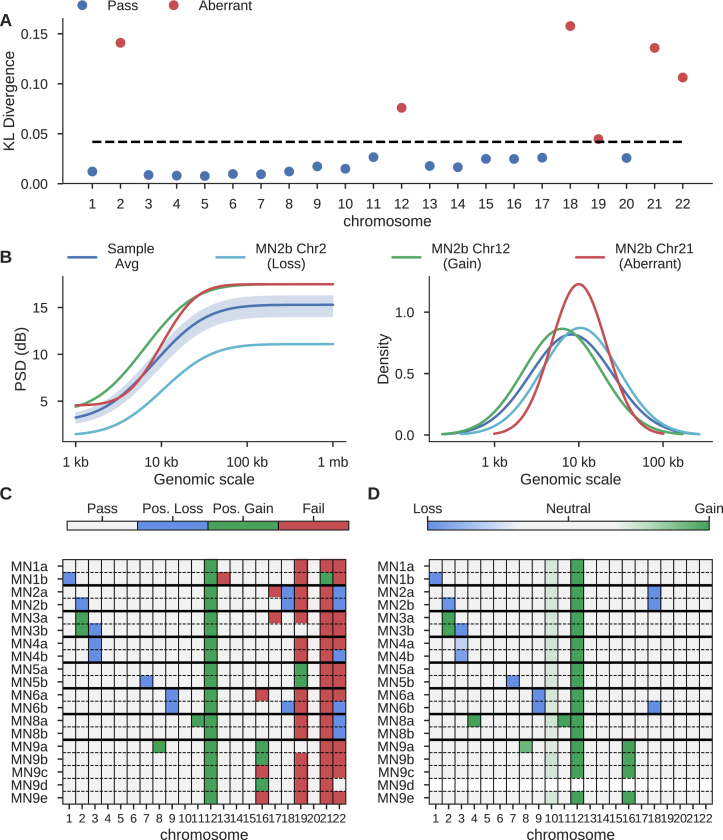
Single cell whole-genome sequencing (scWGS) is providing novel insights into the nature of genetic heterogeneity in normal and diseased cells. However, the whole-genome amplification process required for scWGS introduces biases into the resulting sequencing that can confound downstream analysis. Here, we present a statistical method, with an accompanying package PaSD-qc (Power Spectral Density-qc), that evaluates the properties and quality of single cell libraries. It uses a modified power spectral density to assess amplification uniformity, amplicon size distribution, autocovariance and inter-sample consistency as well as to identify chromosomes with aberrant read-density profiles due either to copy alterations or poor amplification. These metrics provide a standard way to compare the quality of single cell samples as well as yield information necessary to improve variant calling strategies. We demonstrate the usefulness of this tool in comparing the properties of scWGS protocols, identifying potential chromosomal copy number variation, determining chromosomal and subchromosomal regions of poor amplification, and selecting high-quality libraries from low-coverage data for deep sequencing. The software is available free and open-source at https://github.com/parklab/PaSDqc.
单细胞全基因组测序(scWGS)为正常和患病细胞中遗传异质性的本质提供了新的见解。然而,scWGS 所需的全基因组扩增过程会给后续的测序分析带来偏差。在这里,我们提出了一种统计方法,同时提供了一个配套的软件包 PaSD-qc(功率谱密度-qc),用于评估单细胞文库的特性和质量。它使用改进的功率谱密度来评估扩增均匀性、扩增子大小分布、自协方差和样本间一致性,以及识别由于拷贝改变或扩增不良而导致异常读密度分布的染色体。这些指标为比较单细胞样本的质量提供了一种标准方法,并提供了必要的信息来改进变异调用策略。我们展示了该工具在比较 scWGS 方案的特性、识别潜在的染色体拷贝数变异、确定染色体和亚染色体扩增不良区域,以及从低覆盖数据中选择高质量文库进行深度测序方面的有用性。该软件可在 https://github.com/parklab/PaSDqc 免费获取并开放源代码。