School of Computer Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore.
School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore, 637551, Singapore.
BMC Bioinformatics. 2020 Apr 16;21(1):147. doi: 10.1186/s12859-020-3480-3.
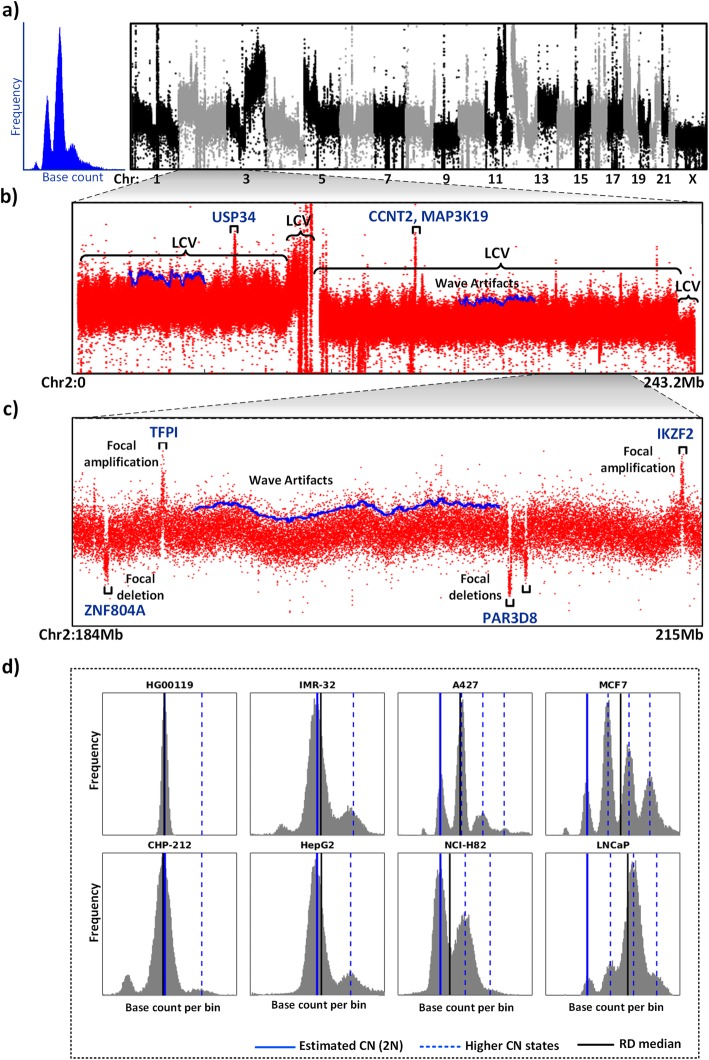
Detection of DNA copy number alterations (CNAs) is critical to understand genetic diversity, genome evolution and pathological conditions such as cancer. Cancer genomes are plagued with widespread multi-level structural aberrations of chromosomes that pose challenges to discover CNAs of different length scales, and distinct biological origins and functions. Although several computational tools are available to identify CNAs using read depth (RD) signal, they fail to distinguish between large-scale and focal alterations due to inaccurate modeling of the RD signal of cancer genomes. Additionally, RD signal is affected by overdispersion-driven biases at low coverage, which significantly inflate false detection of CNA regions.
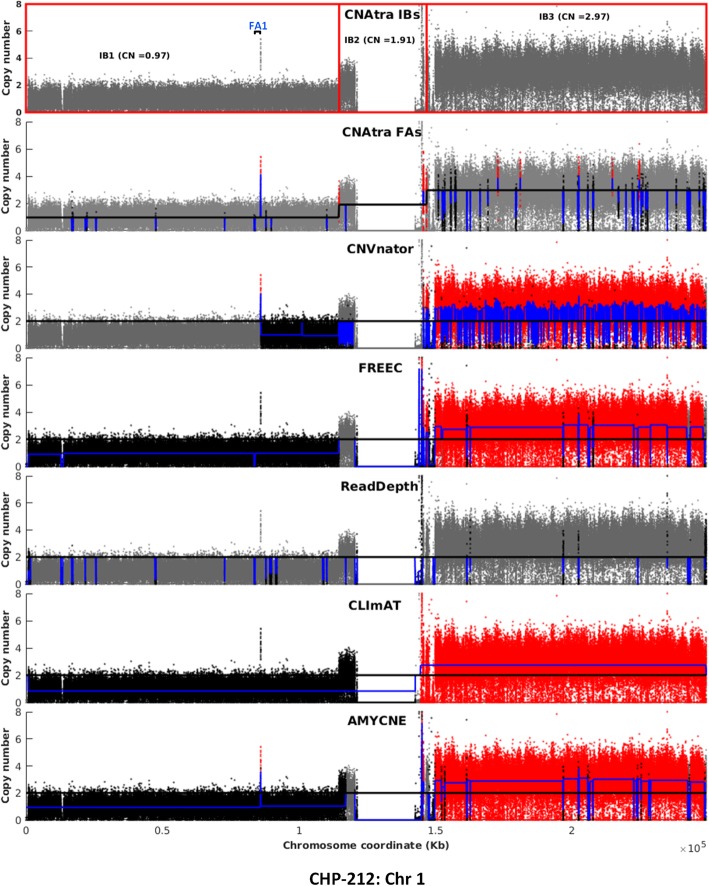
We have developed CNAtra framework to hierarchically discover and classify 'large-scale' and 'focal' copy number gain/loss from a single whole-genome sequencing (WGS) sample. CNAtra first utilizes a multimodal-based distribution to estimate the copy number (CN) reference from the complex RD profile of the cancer genome. We implemented Savitzky-Golay smoothing filter and Modified Varri segmentation to capture the change points of the RD signal. We then developed a CN state-driven merging algorithm to identify the large segments with distinct copy numbers. Next, we identified focal alterations in each large segment using coverage-based thresholding to mitigate the adverse effects of signal variations. Using cancer cell lines and patient datasets, we confirmed CNAtra's ability to detect and distinguish the segmental aneuploidies and focal alterations. We used realistic simulated data for benchmarking the performance of CNAtra against other single-sample detection tools, where we artificially introduced CNAs in the original cancer profiles. We found that CNAtra is superior in terms of precision, recall and f-measure. CNAtra shows the highest sensitivity of 93 and 97% for detecting large-scale and focal alterations respectively. Visual inspection of CNAs revealed that CNAtra is the most robust detection tool for low-coverage cancer data.
CNAtra is a single-sample CNA detection tool that provides an analytical and visualization framework for CNA profiling without relying on any reference control. It can detect chromosome-level segmental aneuploidies and high-confidence focal alterations, even from low-coverage data. CNAtra is an open-source software implemented in MATLAB. It is freely available at https://github.com/AISKhalil/CNAtra.
检测 DNA 拷贝数改变(CNAs)对于理解遗传多样性、基因组进化以及癌症等病理状况至关重要。癌症基因组中广泛存在染色体的多水平结构异常,这给发现不同长度尺度、不同起源和功能的 CNA 带来了挑战。虽然有几种计算工具可用于使用读取深度(RD)信号来识别 CNA,但由于对癌症基因组 RD 信号的建模不准确,它们无法区分大尺度和局灶性改变。此外,RD 信号会受到低覆盖下过度分散驱动的偏差的影响,这会显著增加 CNA 区域的假阳性检测。
我们开发了 CNAtra 框架,用于从单个全基因组测序(WGS)样本中分层发现和分类“大尺度”和“局灶性”拷贝数增益/丢失。CNAtra 首先利用多模态分布从癌症基因组复杂的 RD 谱中估计拷贝数(CN)参考。我们实现了 Savitzky-Golay 平滑滤波器和 Modified Varri 分段来捕获 RD 信号的变化点。然后,我们开发了一种基于 CN 状态的合并算法来识别具有不同拷贝数的大段。接下来,我们使用基于覆盖的阈值在每个大段中识别局灶性改变,以减轻信号变化的不利影响。使用癌细胞系和患者数据集,我们证实了 CNAtra 检测和区分片段非整倍性和局灶性改变的能力。我们使用真实模拟数据来评估 CNAtra 与其他单样本检测工具的性能,在这些工具中,我们在原始癌症图谱中人为引入了 CNA。我们发现,CNAtra 在精度、召回率和 F1 度量方面表现更优。CNAtra 分别在检测大尺度和局灶性改变方面具有 93%和 97%的最高灵敏度。对 CNA 的可视化检查表明,CNAtra 是用于低覆盖癌症数据的最稳健检测工具。
CNAtra 是一种单样本 CNA 检测工具,它提供了一个用于 CNA 分析和可视化的框架,无需依赖任何参考控制。它可以检测染色体水平的片段非整倍性和高置信度的局灶性改变,即使来自低覆盖数据。CNAtra 是一个用 MATLAB 实现的开源软件。它可在 https://github.com/AISKhalil/CNAtra 上免费获取。