Cong Yalong, Li Mengxin, Feng Guoqiang, Li Yuchen, Wang Xianwei, Duan Lili
Shandong Province Key Laboratory of Medical Physics and Image Processing Technology, School of Physics and Electronics, Shandong Normal University, Jinan, 250014, China.
Center for Optics & Optoelectronics Research, College of Science, Zhejiang University of Technology, Hangzhou, 310023, China.
Sci Rep. 2017 Dec 18;7(1):17708. doi: 10.1038/s41598-017-17868-z.
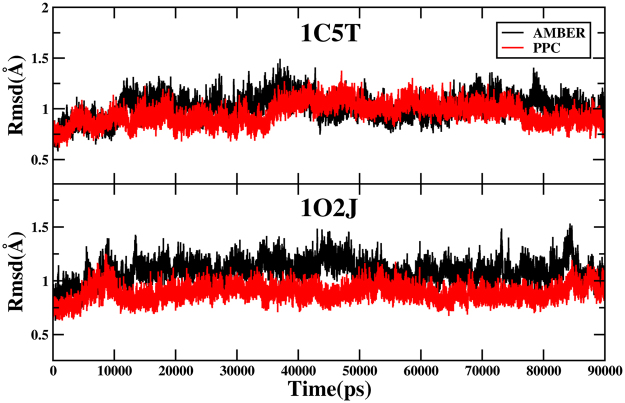
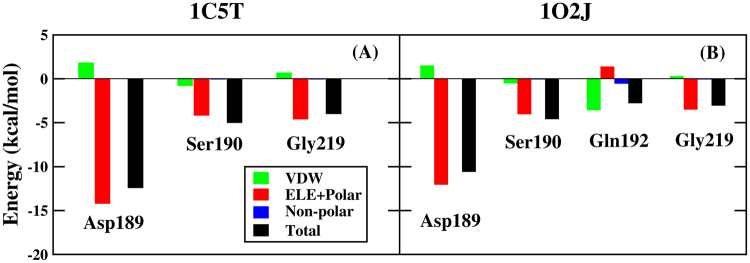
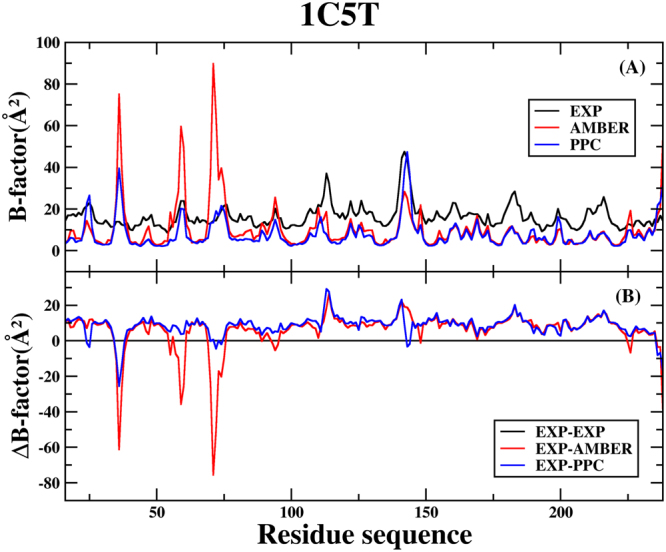
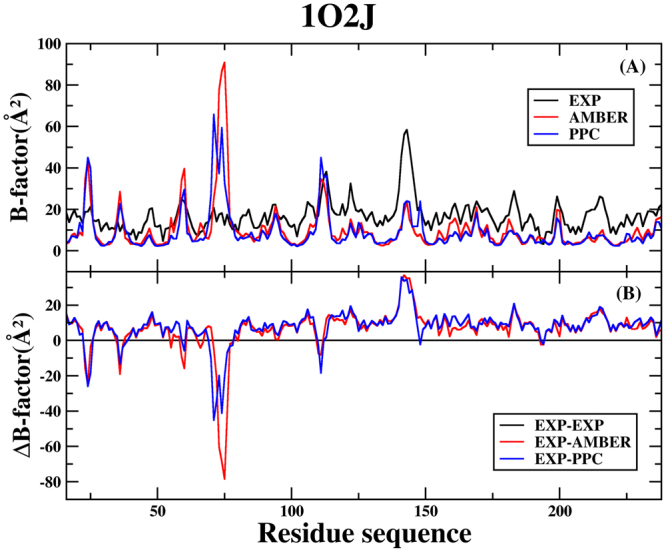
Molecular dynamics (MD) simulation in the explicit water is performed to study the interaction mechanism of trypsin-ligand binding under the AMBER force field and polarized protein-specific charge (PPC) force field combined the new developed highly efficient interaction entropy (IE) method for calculation of entropy change. And the detailed analysis and comparison of the results of MD simulation for two trypsin-ligand systems show that the root-mean-square deviation (RMSD) of backbone atoms, B-factor, intra-protein and protein-ligand hydrogen bonds are more stable under PPC force field than AMBER force field. Our results demonstrate that the IE method is superior than the traditional normal mode (Nmode) method in the calculation of entropy change and the calculated binding free energy under the PPC force field combined with the IE method is more close to the experimental value than other three combinations (AMBER-Nmode, AMBER-IE and PPC-Nmode). And three critical hydrogen bonds between trypsin and ligand are broken under AMBER force field. However, they are well preserved under PPC force field. Detailed binding interactions of ligands with trypsin are further analyzed. The present work demonstrates that the polarized force field combined the highly efficient IE method is critical in MD simulation and free energy calculation.
在显式水环境中进行分子动力学(MD)模拟,以研究在AMBER力场和极化蛋白特异性电荷(PPC)力场下胰蛋白酶-配体结合的相互作用机制,并结合新开发的高效相互作用熵(IE)方法来计算熵变。对两个胰蛋白酶-配体系统的MD模拟结果进行详细分析和比较表明,与AMBER力场相比,PPC力场下主链原子的均方根偏差(RMSD)、B因子、蛋白内和蛋白-配体氢键更稳定。我们的结果表明,在熵变计算中,IE方法优于传统的简正模式(Nmode)方法,并且与其他三种组合(AMBER-Nmode、AMBER-IE和PPC-Nmode)相比,结合IE方法的PPC力场下计算得到的结合自由能更接近实验值。在AMBER力场下,胰蛋白酶与配体之间的三个关键氢键断裂。然而,在PPC力场下它们得到了很好的保留。进一步分析了配体与胰蛋白酶的详细结合相互作用。目前的工作表明,极化力场与高效IE方法相结合在MD模拟和自由能计算中至关重要。