Feng Ting, Chen Fu, Kang Yu, Sun Huiyong, Liu Hui, Li Dan, Zhu Feng, Hou Tingjun
College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, Zhejiang, China.
State Key Lab of CAD&CG, Zhejiang University, Hangzhou, 310058, Zhejiang, China.
J Cheminform. 2017 Dec 28;9(1):66. doi: 10.1186/s13321-017-0254-7.
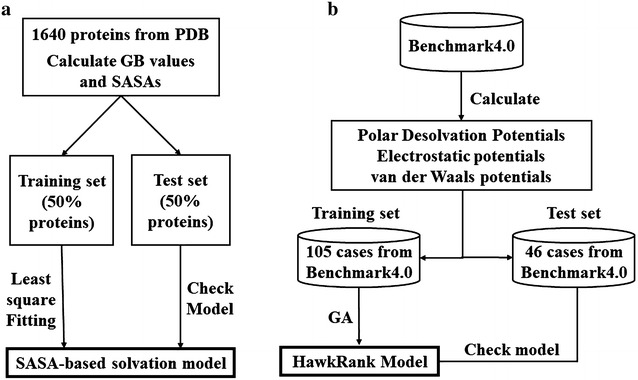
Deciphering the structural determinants of protein-protein interactions (PPIs) is essential to gain a deep understanding of many important biological functions in the living cells. Computational approaches for the structural modeling of PPIs, such as protein-protein docking, are quite needed to complement existing experimental techniques. The reliability of a protein-protein docking method is dependent on the ability of the scoring function to accurately distinguish the near-native binding structures from a huge number of decoys. In this study, we developed HawkRank, a novel scoring function designed for the sampling stage of protein-protein docking by summing the contributions from several energy terms, including van der Waals potentials, electrostatic potentials and desolvation potentials. First, based on the solvation free energies predicted by the Generalized Born model for ~ 800 proteins, a SASA (solvent accessible surface area)-based solvation model was developed, which can give the aqueous solvation free energies for proteins by summing the contributions of 21 atom types. Then, the van der Waals potentials and electrostatic potentials based on the Amber ff14SB force field were computed. Finally, the HawkRank scoring function was derived by determining the most optimal weights for five energy terms based on the training set. Here, MSR (modified success rate), a novel protein-protein scoring quality index, was used to assess the performance of HawkRank and three other popular protein-protein scoring functions, including ZRANK, FireDock and dDFIRE. The results show that HawkRank outperformed the other three scoring functions according to the total number of hits and MSR. HawkRank is available at http://cadd.zju.edu.cn/programs/hawkrank .
解析蛋白质-蛋白质相互作用(PPI)的结构决定因素对于深入理解活细胞中许多重要的生物学功能至关重要。蛋白质-蛋白质对接等用于PPI结构建模的计算方法对于补充现有实验技术非常必要。蛋白质-蛋白质对接方法的可靠性取决于评分函数从大量诱饵中准确区分近天然结合结构的能力。在本研究中,我们开发了HawkRank,这是一种新颖的评分函数,通过将包括范德华势、静电势和去溶剂化势在内的几个能量项的贡献相加,专为蛋白质-蛋白质对接的采样阶段而设计。首先,基于广义玻恩模型预测的约800种蛋白质的溶剂化自由能,开发了一种基于溶剂可及表面积(SASA)的溶剂化模型,该模型可以通过对21种原子类型的贡献求和来给出蛋白质的水相溶剂化自由能。然后,计算基于Amber ff14SB力场的范德华势和静电势。最后,通过基于训练集确定五个能量项的最优权重得出HawkRank评分函数。在此,使用一种新颖的蛋白质-蛋白质评分质量指标MSR(修正成功率)来评估HawkRank和其他三种流行的蛋白质-蛋白质评分函数(包括ZRANK、FireDock和dDFIRE)的性能。结果表明,根据命中总数和MSR,HawkRank优于其他三种评分函数。可在http://cadd.zju.edu.cn/programs/hawkrank获取HawkRank。