Whittaker Gary R, André Nicole M, Millet Jean Kaoru
Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, New York, USA.
mSphere. 2018 Jan 3;3(1). doi: 10.1128/mSphereDirect.00463-17. eCollection 2018 Jan-Feb.
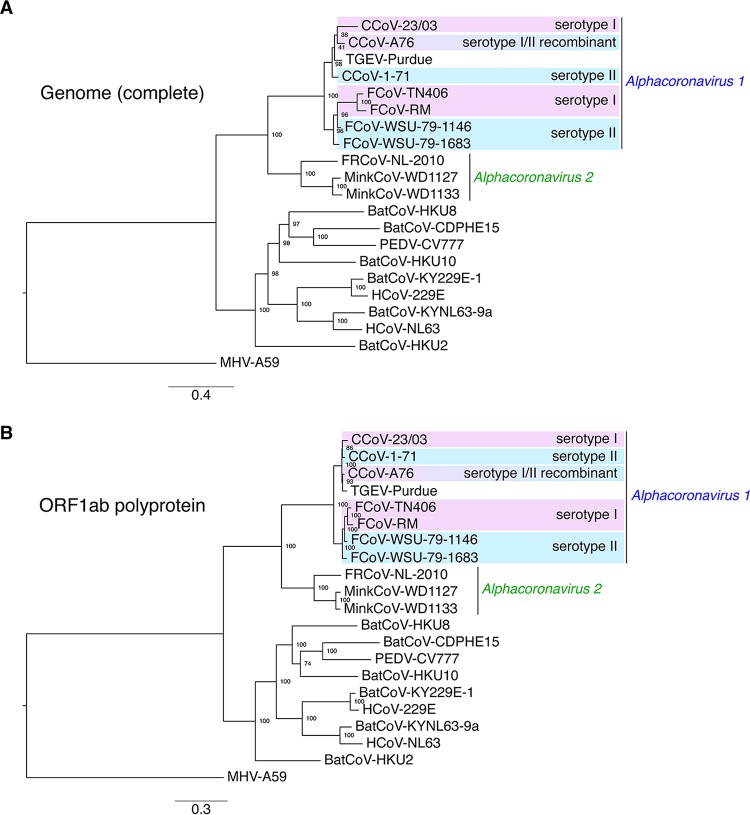
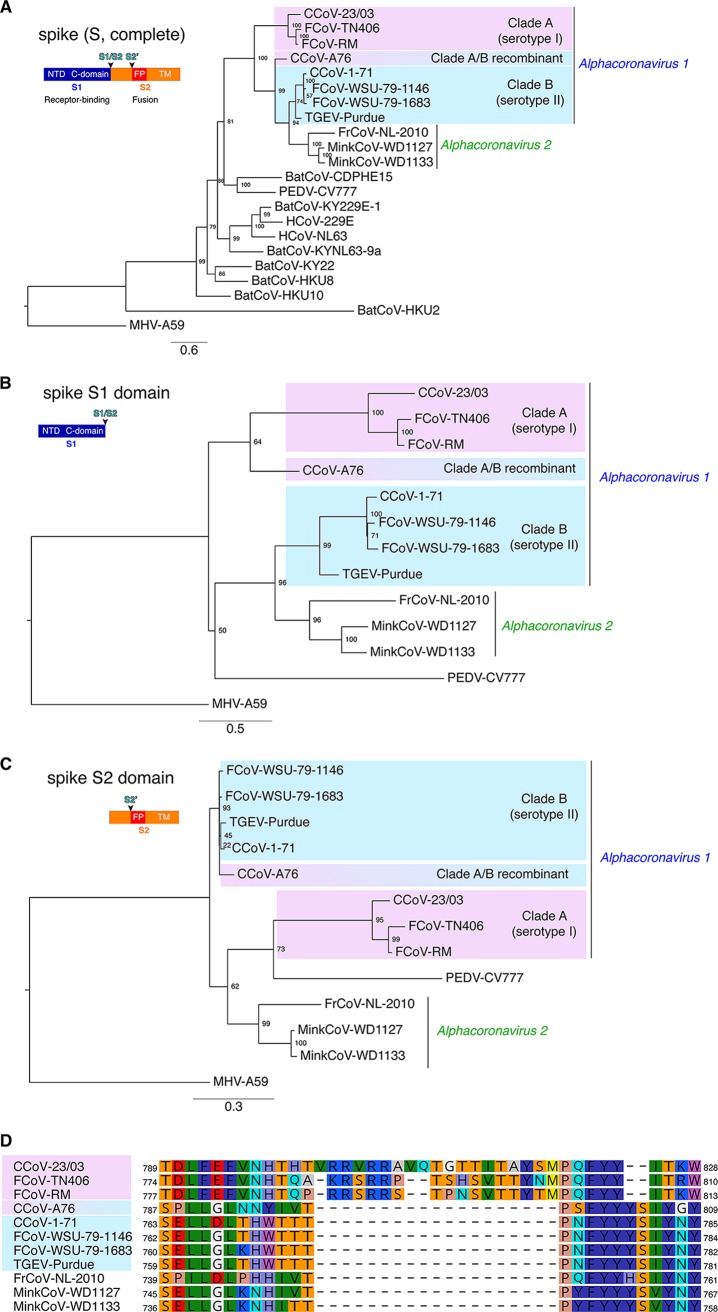
The difficulties related to virus taxonomy have been amplified by recent advances in next-generation sequencing and metagenomics, prompting the field to revisit the question of what constitutes a useful viral classification. Here, taking a challenging classification found in coronaviruses, we argue that consideration of biological properties in addition to sequence-based demarcations is critical for generating useful taxonomy that recapitulates complex evolutionary histories. Within the genus, the species encompasses several biologically distinct viruses. We carried out functionally based phylogenetic analysis, centered on the spike gene, which encodes the main surface antigen and primary driver of tropism and pathogenesis. Within the species, we identify clade A (encompassing serotype I feline coronavirus [FCoV] and canine coronavirus [CCoV]) and clade B (grouping serotype II FCoV and CCoV and transmissible gastroenteritis virus [TGEV]-like viruses). We propose this clade designation, along with the newly proposed species, as an improved way to classify the genus. Our work focuses on improving the classification of the genus. The species groups viruses of veterinary importance that infect distinct mammalian hosts and includes canine and feline coronaviruses and transmissible gastroenteritis virus. It is the prototype species of the genus; however, it encompasses biologically distinct viruses. To better characterize this prototypical species, we performed phylogenetic analyses based on the sequences of the spike protein, one of the main determinants of tropism and pathogenesis, and reveal the existence of two subgroups or clades that fit with previously established serotype demarcations. We propose a new clade designation to better classify members.
下一代测序和宏基因组学的最新进展加剧了与病毒分类学相关的困难,促使该领域重新审视什么构成有用的病毒分类这一问题。在此,以冠状病毒中一个具有挑战性的分类为例,我们认为除了基于序列的划分外,考虑生物学特性对于生成能够概括复杂进化历史的有用分类学至关重要。在该属内,该种包含几种生物学上不同的病毒。我们以编码主要表面抗原以及嗜性和发病机制的主要驱动因素的刺突基因为中心,进行了基于功能的系统发育分析。在该种内,我们确定了A分支(包括I型猫冠状病毒[FCoV]和犬冠状病毒[CCoV])和B分支(将II型FCoV和CCoV以及传染性胃肠炎病毒[TGEV]样病毒归为一组)。我们提议这种分支命名以及新提议的种,作为对该属进行分类的一种改进方法。我们的工作重点是改进该属的分类。该种将感染不同哺乳动物宿主的具有兽医重要性的病毒归为一组,包括犬冠状病毒和猫冠状病毒以及传染性胃肠炎病毒。它是该属的原型种;然而,它包含生物学上不同的病毒。为了更好地表征这个原型种,我们基于刺突蛋白的序列进行了系统发育分析,刺突蛋白是嗜性和发病机制的主要决定因素之一,并揭示了存在两个与先前确定的血清型划分相符的亚组或分支。我们提议一种新的分支命名以更好地对该属成员进行分类。