Sundriyal Sandeep, Chen Patty B, Lubin Alexandra S, Lueg Gregor A, Li Fengling, White Andrew J P, Malmquist Nicholas A, Vedadi Masoud, Scherf Artur, Fuchter Matthew J
Department of Chemistry , Imperial College London , London SW7 2AZ , UK . Email:
Unité Biologie des Interactions Hôte-Parasite , Département de Parasites et Insectes Vecteurs , Institut Pasteur , Paris 75015 , France.
Medchemcomm. 2017 May 1;8(5):1069-1092. doi: 10.1039/c7md00052a. Epub 2017 Mar 15.
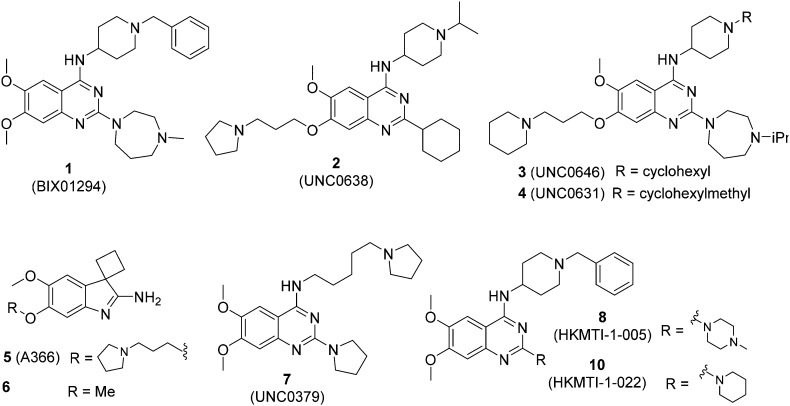
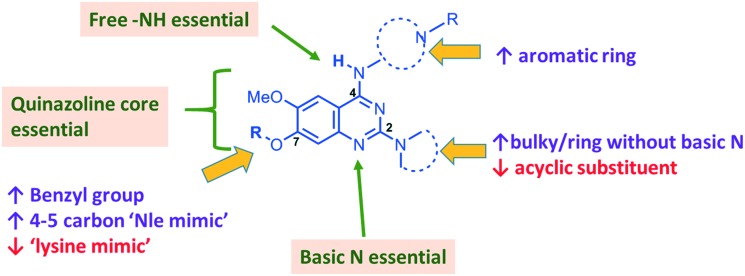
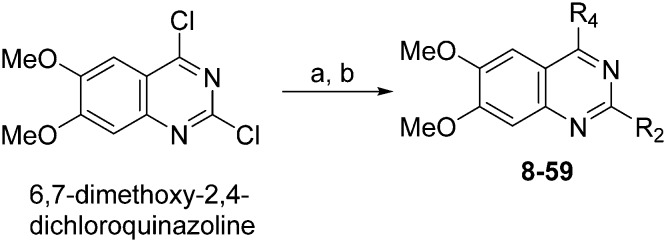
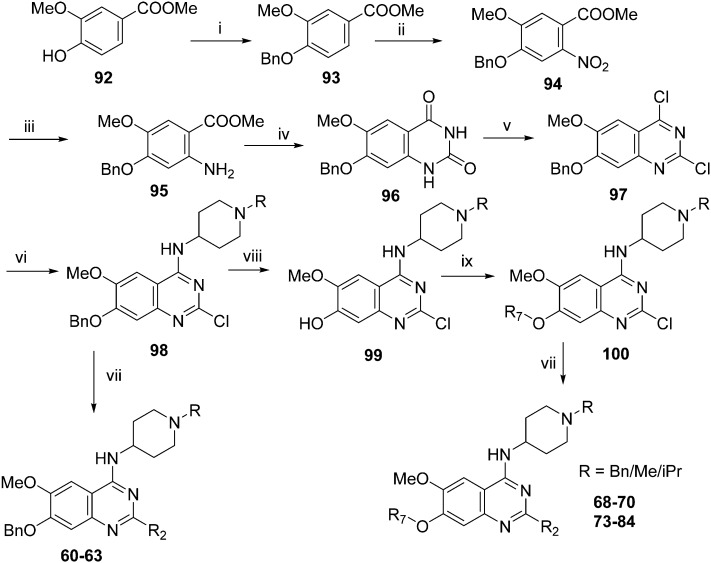
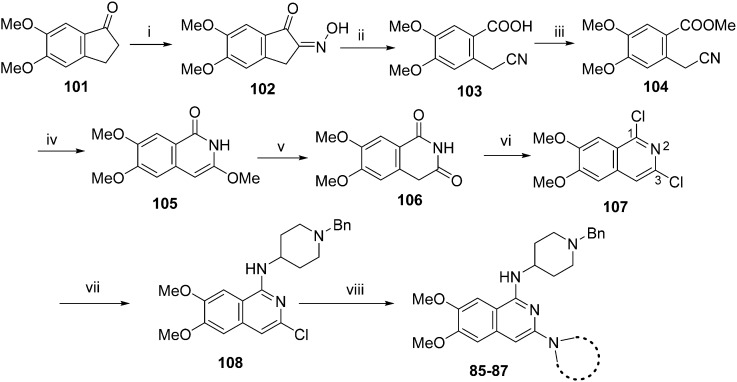
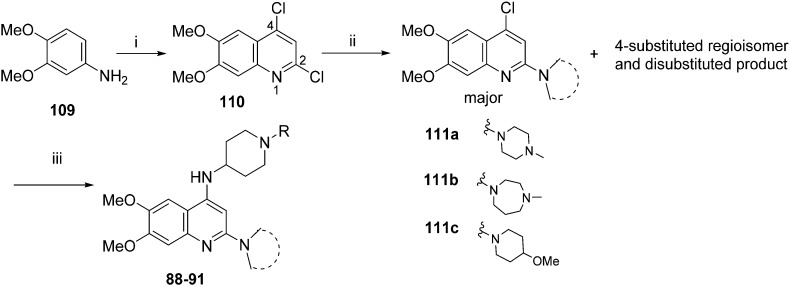
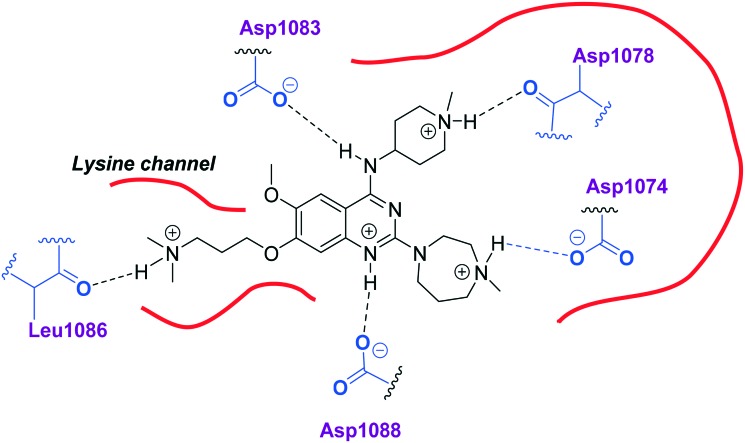
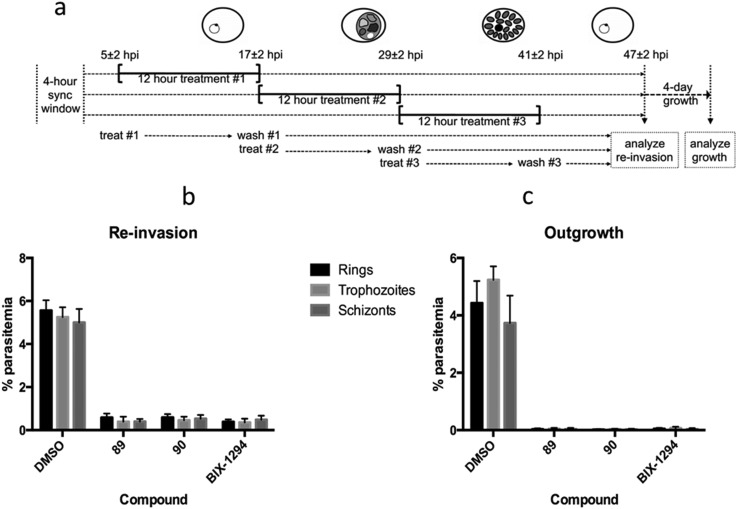
HKMTs (PfHKMTs) play a key role in controlling gene expression and represent exciting new anti-malarial epigenetic targets. Using an inhibitor series derived from the diaminoquinazoline HKMT inhibitory chemotype, we have previously identified compounds with highly promising antimalarial activity, including irreversible asexual cycle blood stage-independent cytotoxic activity at nM concentrations, oral efficacy in models of disease, and the unprecedented ability to reactivate dormant liver stage parasites (hypnozoites). However, future development of this series will need to address host parasite selectivity, where inhibitory activity against human G9a is removed from the lead compounds, while maintaining potent anti- activity. Herein, we report an extensive study of the SAR of this series against both G9a and . We have identified key SAR features which demonstrate that high parasite G9a selectivity can be achieved by selecting appropriate substituents at position 2, 4 and 7 of the quinazoline ring. We have also, in turn, discovered that potent G9a inhibitors can be identified by employing a 6-carbon 'Nle mimic' at position 7. Together, this data suggests that while broadly similar, the G9a and potential PfHKMT target(s) binding pockets and/or binding modes of the diaminoquinazoline analogues exhibit clear and exploitable differences. Based on this, we believe this scaffold to have clear potential for development into a novel anti-malarial therapeutic.
组蛋白赖氨酸甲基转移酶(PfHKMTs)在控制基因表达中起关键作用,是令人兴奋的新型抗疟疾表观遗传靶点。利用源自二氨基喹唑啉HKMT抑制化学类型的一系列抑制剂,我们先前已鉴定出具有极有前景的抗疟活性的化合物,包括在纳摩尔浓度下具有不可逆的无性周期血期非依赖性细胞毒性活性、在疾病模型中的口服疗效以及重新激活休眠肝期寄生虫(休眠子)的前所未有的能力。然而,该系列化合物的未来开发需要解决宿主-寄生虫选择性问题,即从先导化合物中去除对人G9a的抑制活性,同时保持强效的抗疟活性。在此,我们报告了该系列化合物针对G9a和疟原虫的构效关系的广泛研究。我们已经确定了关键的构效关系特征,表明通过在喹唑啉环的2、4和7位选择合适的取代基,可以实现高疟原虫-G9a选择性。反过来,我们还发现通过在7位使用6碳的“Nle模拟物”可以鉴定出强效的G9a抑制剂。总之,这些数据表明,虽然二氨基喹唑啉类似物的G9a和潜在的PfHKMT靶点结合口袋和/或结合模式大致相似,但它们表现出明显且可利用的差异。基于此,我们认为该骨架具有开发成新型抗疟疾治疗药物的明显潜力。