Newey Paul J, Berg Jonathan N, Zhou Kaixin, Palmer Colin N A, Thakker Rajesh V
Division of Molecular & Clinical Medicine, Ninewells Hospital & Medical School, University of Dundee, Dundee, DD1 9SY, Scotland, United Kingdom.
Clinical Genetics, Ninewells Hospital & Medical School, University of Dundee, Dundee, DD1 9SY Scotland, United Kingdom.
J Endocr Soc. 2017 Nov 15;1(12):1507-1526. doi: 10.1210/js.2017-00330. eCollection 2017 Dec 1.
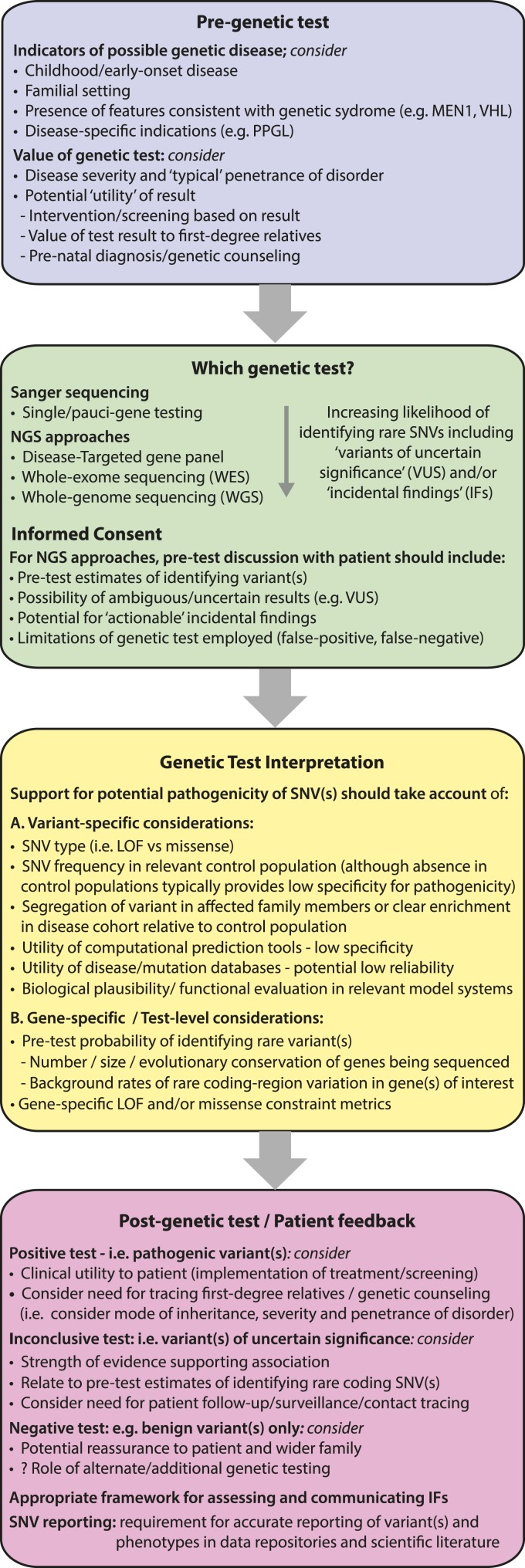
Genetic testing is increasingly used for clinical diagnosis, although variant interpretation presents a major challenge because of high background rates of rare coding-region variation, which may contribute to inaccurate estimates of variant pathogenicity and disease penetrance.
To use the Exome Aggregation Consortium (ExAC) data set to determine the background population frequencies of rare germline coding-region variants in genes associated with hereditary endocrine disease and to evaluate the clinical utility of these data.
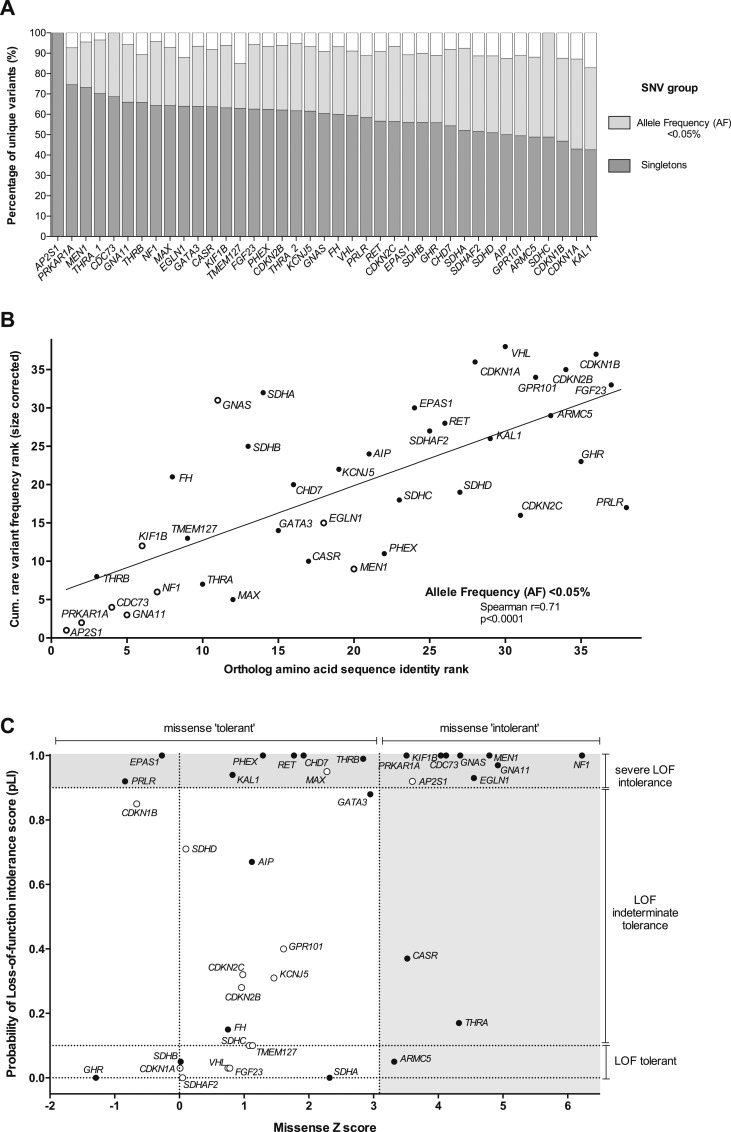
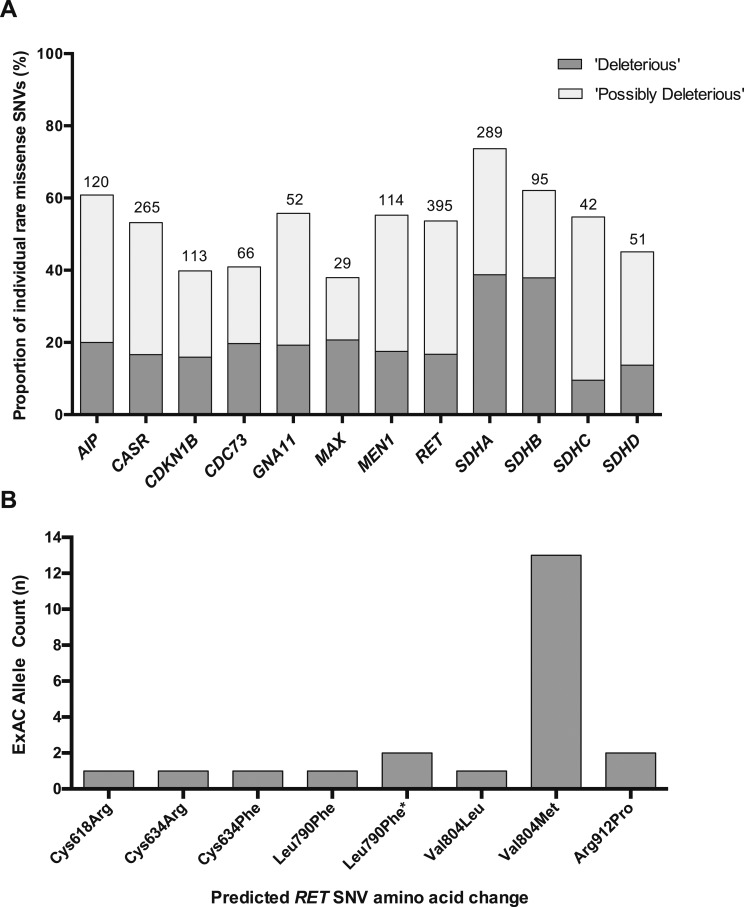
Cumulative frequencies of rare nonsynonymous single-nucleotide variants were established for 38 endocrine disease genes in 60,706 unrelated control individuals. The utility of gene-level and variant-level metrics of tolerability was assessed, and the pathogenicity and penetrance of germline variants previously associated with endocrine disease evaluated.
The frequency of rare coding-region variants differed markedly between genes and was correlated with the degree of evolutionary conservation. Genes associated with dominant monogenic endocrine disorders typically harbored fewer rare missense and/or loss-of-function variants than expected. variant prediction tools demonstrated low clinical specificity. The frequency of several endocrine disease‒associated variants in the ExAC cohort far exceeded estimates of disease prevalence, indicating either misclassification or overestimation of disease penetrance. Finally, we illustrate how rare variant frequencies may be used to anticipate expected rates of background rare variation when performing disease-targeted genetic testing.
Quantifying the frequency and spectrum of rare variation using population-level sequence data facilitates improved estimates of variant pathogenicity and penetrance and should be incorporated into the clinical decision-making algorithm when undertaking genetic testing.
基因检测越来越多地用于临床诊断,尽管由于罕见编码区变异的背景率较高,变异解读面临重大挑战,这可能导致对变异致病性和疾病外显率的估计不准确。
利用外显子聚合联盟(ExAC)数据集确定与遗传性内分泌疾病相关基因中罕见种系编码区变异的背景人群频率,并评估这些数据的临床实用性。
设计、设置、参与者:在60706名无亲缘关系的对照个体中,确定了38个内分泌疾病基因的罕见非同义单核苷酸变异的累积频率。评估了基因水平和变异水平耐受性指标的实用性,并评估了先前与内分泌疾病相关的种系变异的致病性和外显率。
罕见编码区变异的频率在不同基因之间差异显著,且与进化保守程度相关。与显性单基因内分泌疾病相关的基因通常携带的罕见错义变异和/或功能丧失变异比预期的少。变异预测工具显示临床特异性较低。ExAC队列中几种与内分泌疾病相关的变异频率远远超过疾病患病率估计值,表明存在疾病分类错误或疾病外显率估计过高的情况。最后,我们说明了在进行疾病靶向基因检测时,如何利用罕见变异频率来预测背景罕见变异的预期发生率。
利用人群水平的序列数据量化罕见变异的频率和谱,有助于改进对变异致病性和外显率的估计,并且在进行基因检测时应纳入临床决策算法中。