Del Castillo Francisco J, Del Castillo Ignacio
Servicio de Genética, Hospital Universitario Ramón y Cajal, IRYCIS, Madrid, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain.
Front Mol Neurosci. 2017 Dec 22;10:428. doi: 10.3389/fnmol.2017.00428. eCollection 2017.
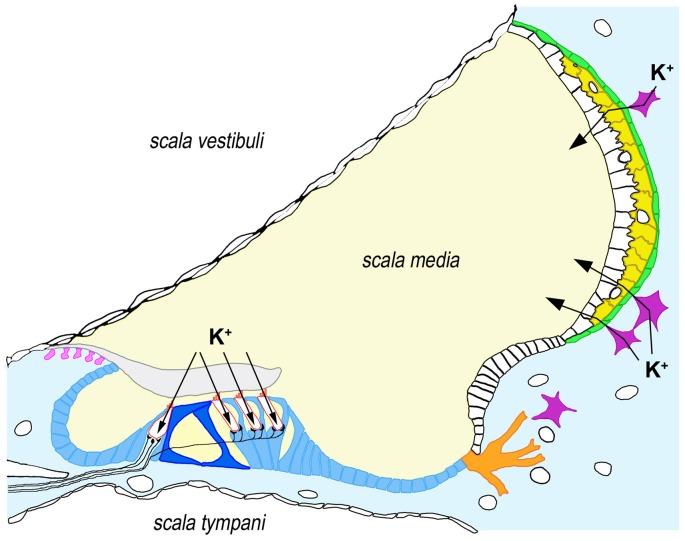
The inner ear is a very complex sensory organ whose development and function depend on finely balanced interactions among diverse cell types. The many different kinds of inner ear supporting cells play the essential roles of providing physical and physiological support to sensory hair cells and of maintaining cochlear homeostasis. Appropriately enough, the gene most commonly mutated among subjects with hereditary hearing impairment (HI), , encodes the connexin-26 (Cx26) gap-junction channel protein that underlies both intercellular communication among supporting cells and homeostasis of the cochlear fluids, endolymph and perilymph. lies at the DFNB1 locus on 13q12. The specific kind of HI associated with this locus is caused by recessively-inherited mutations that inactivate the two alleles of the gene, either in homozygous or compound heterozygous states. We describe the many diverse classes of genetic alterations that result in DFNB1 HI, such as large deletions that either destroy the gene or remove a regulatory element essential for expression, point mutations that interfere with promoter function or splicing, and small insertions or deletions and nucleotide substitutions that target the coding sequence. We focus on how these alterations disrupt and Cx26 functions and on their different effects on cochlear development and physiology. We finally discuss the diversity of clinical features of DFNB1 HI as regards severity, age of onset, inner ear malformations and vestibular dysfunction, highlighting the areas where future research should be concentrated.
内耳是一个非常复杂的感觉器官,其发育和功能依赖于多种细胞类型之间精确平衡的相互作用。内耳中许多不同类型的支持细胞发挥着重要作用,为感觉毛细胞提供物理和生理支持,并维持耳蜗内环境稳定。恰如其分的是,在遗传性听力损失(HI)患者中最常发生突变的基因,编码连接蛋白26(Cx26)间隙连接通道蛋白,该蛋白是支持细胞间细胞通讯以及耳蜗内淋巴液和外淋巴液内环境稳定的基础。 位于13q12的DFNB1位点。与该位点相关的特定类型的HI是由隐性遗传突变引起的,这些突变使该基因的两个等位基因失活,呈纯合或复合杂合状态。我们描述了导致DFNB1 HI的多种不同类型的基因改变,例如破坏该基因或去除其表达所必需的调控元件的大片段缺失、干扰启动子功能或剪接的点突变,以及靶向该基因编码序列的小插入或缺失和核苷酸替换。我们重点关注这些改变如何破坏该基因和Cx26的功能,以及它们对耳蜗发育和生理学的不同影响。我们最后讨论了DFNB1 HI在严重程度、发病年龄、内耳畸形和前庭功能障碍方面临床特征的多样性,强调了未来研究应集中的领域。