Computational Biology and Bioinformatics Program, Departments of Biological Sciences, Chemistry, Physics & Astronomy, and Computer Science, University of Southern California, Los Angeles, California 90089, USA.
Genome Res. 2018 Mar 1;28(3):321-333. doi: 10.1101/gr.220079.116.
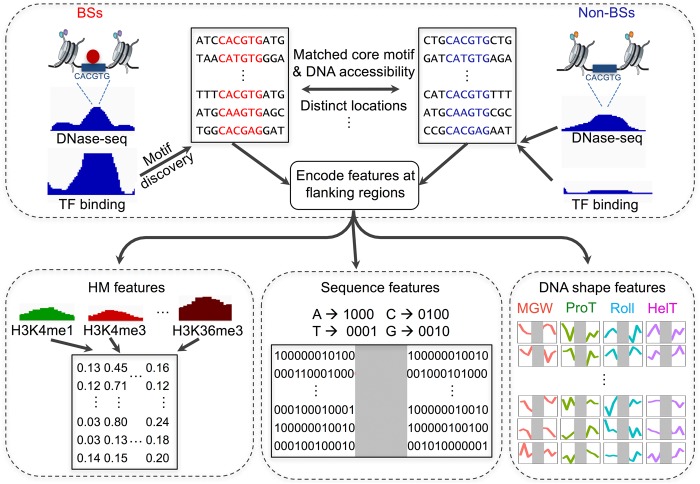
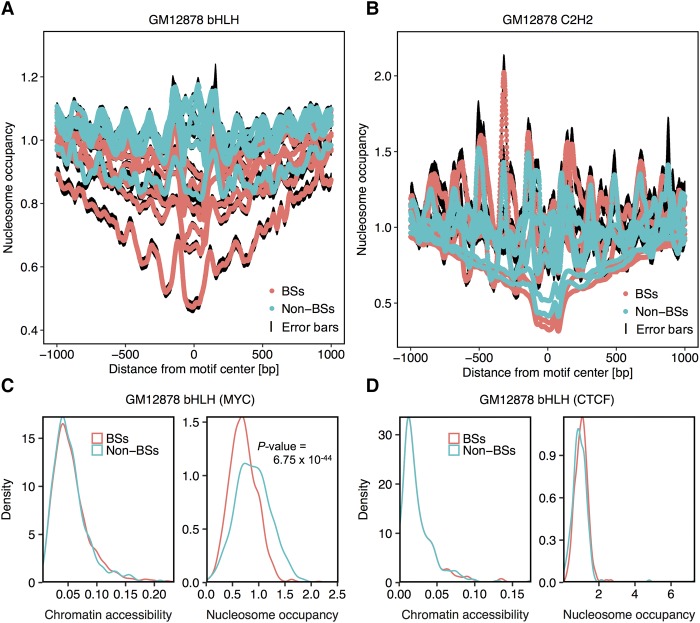
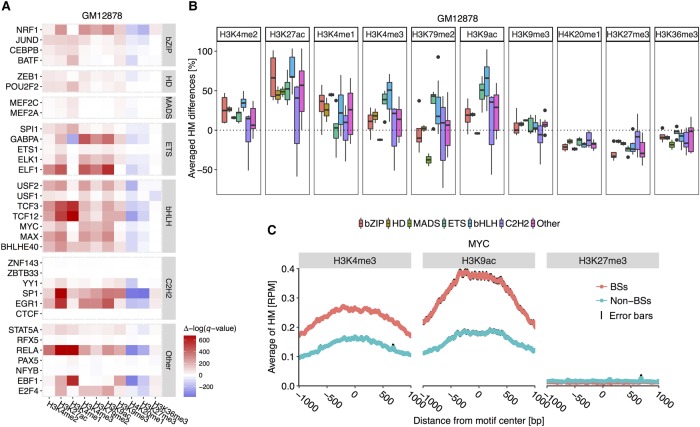
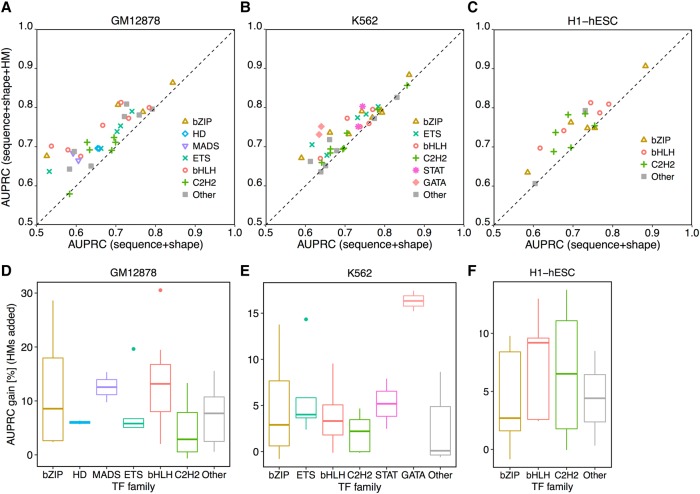
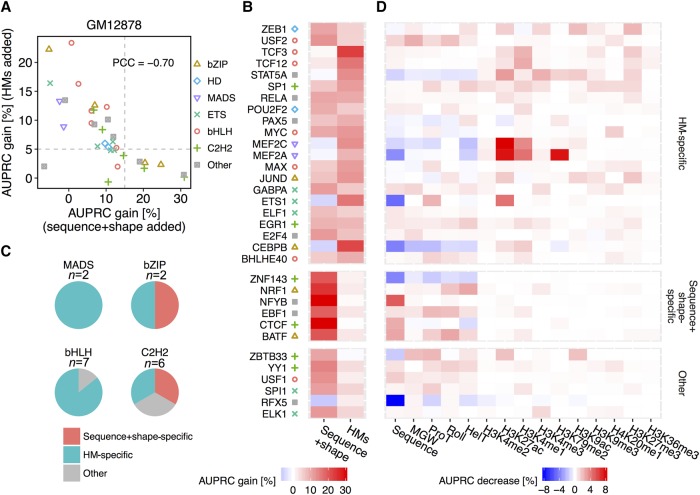
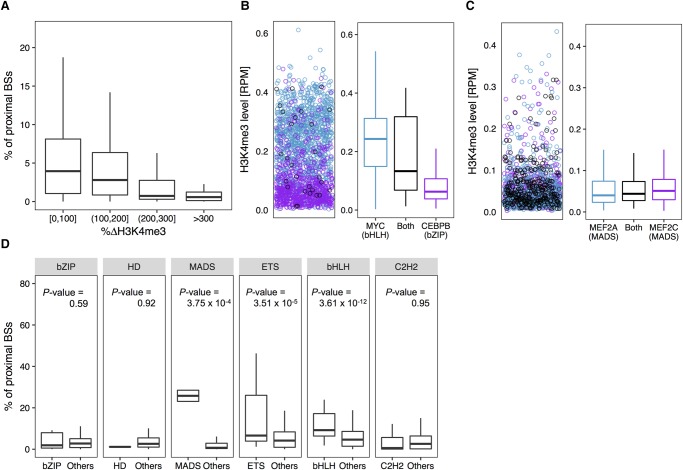
The very small fraction of putative binding sites (BSs) that are occupied by transcription factors (TFs) in vivo can be highly variable across different cell types. This observation has been partly attributed to changes in chromatin accessibility and histone modification (HM) patterns surrounding BSs. Previous studies focusing on BSs within DNA regulatory regions found correlations between HM patterns and TF binding specificities. However, a mechanistic understanding of TF-DNA binding specificity determinants is still not available. The ability to predict in vivo TF binding on a genome-wide scale requires the identification of features that determine TF binding based on evolutionary relationships of DNA binding proteins. To reveal protein family-dependent mechanisms of TF binding, we conducted comprehensive comparisons of HM patterns surrounding BSs and non-BSs with exactly matched core motifs for TFs in three cell lines: 33 TFs in GM12878, 37 TFs in K562, and 18 TFs in H1-hESC. These TFs displayed protein family-specific preferences for HM patterns surrounding BSs, with high agreement among cell lines. Moreover, compared to models based on DNA sequence and shape at flanking regions of BSs, HM-augmented quantitative machine-learning methods resulted in increased performance in a TF family-specific manner. Analysis of the relative importance of features in these models indicated that TFs, displaying larger HM pattern differences between BSs and non-BSs, bound DNA in an HM-specific manner on a protein family-specific basis. We propose that TF family-specific HM preferences reveal distinct mechanisms that assist in guiding TFs to their cognate BSs by altering chromatin structure and accessibility.
在体内,转录因子(TF)占据的假定结合位点(BS)的很小一部分在不同的细胞类型中可能高度可变。这种观察结果部分归因于 BS 周围染色质可及性和组蛋白修饰(HM)模式的变化。以前专注于 DNA 调控区域内 BS 的研究发现,HM 模式与 TF 结合特异性之间存在相关性。然而,TF-DNA 结合特异性决定因素的机制理解仍然不可用。在全基因组范围内预测 TF 结合的能力需要根据 DNA 结合蛋白的进化关系确定决定 TF 结合的特征。为了揭示 TF 结合的蛋白家族依赖性机制,我们在三个细胞系(GM12878 中的 33 个 TF、K562 中的 37 个 TF 和 H1-hESC 中的 18 个 TF)中对 BS 和具有完全匹配核心基序的非 BS 周围的 HM 模式进行了全面比较。这些 TF 对 BS 周围的 HM 模式表现出蛋白家族特异性偏好,细胞系之间具有高度一致性。此外,与基于 BS 侧翼区域的 DNA 序列和形状的模型相比,HM 增强的定量机器学习方法以 TF 家族特异性的方式提高了性能。这些模型中特征的相对重要性分析表明,显示 BS 和非 BS 之间 HM 模式差异较大的 TF 以 HM 特异性的方式在蛋白家族特异性的基础上结合 DNA。我们提出,TF 家族特异性的 HM 偏好揭示了通过改变染色质结构和可及性来帮助 TF 与其同源 BS 结合的不同机制。