Roy Sunetra, Tomaszowski Karl-Heinz, Luzwick Jessica W, Park Soyoung, Li Jun, Murphy Maureen, Schlacher Katharina
Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston, United States.
Department of Genomic Medicine, University of Texas MD Anderson Cancer Center, Houston, United States.
Elife. 2018 Jan 15;7:e31723. doi: 10.7554/eLife.31723.
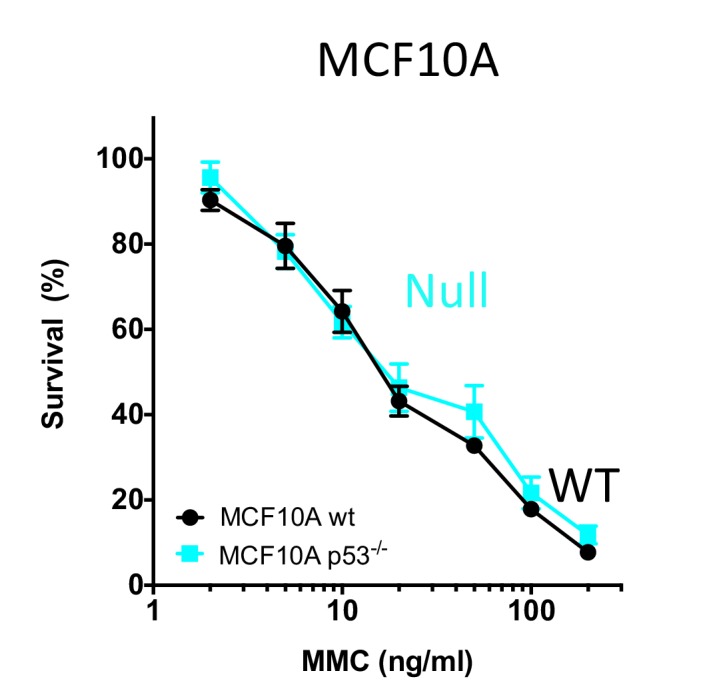
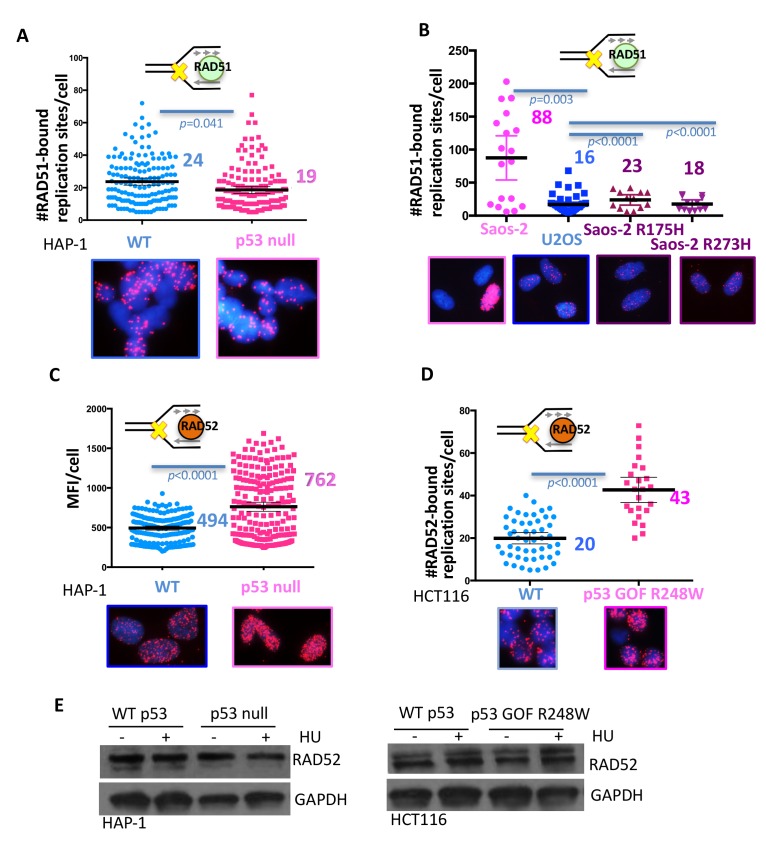
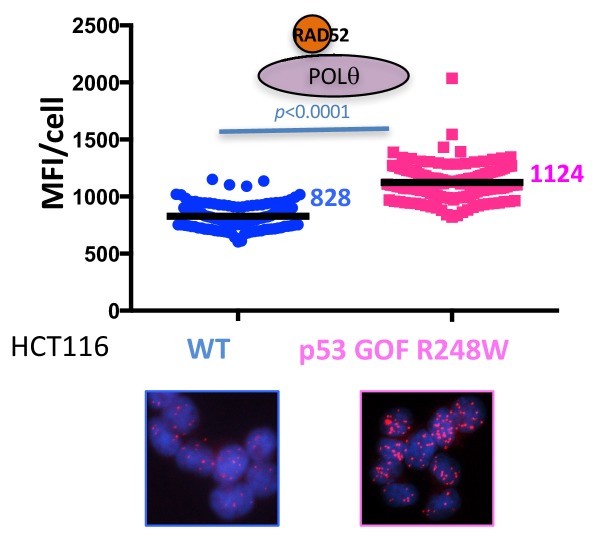
Classically, p53 tumor suppressor acts in transcription, apoptosis, and cell cycle arrest. Yet, replication-mediated genomic instability is integral to oncogenesis, and p53 mutations promote tumor progression and drug-resistance. By delineating human and murine separation-of-function p53 alleles, we find that p53 null and gain-of-function (GOF) mutations exhibit defects in restart of stalled or damaged DNA replication forks that drive genomic instability, which isgenetically separable from transcription activation. By assaying protein-DNA fork interactions in single cells, we unveil a p53-MLL3-enabled recruitment of MRE11 DNA replication restart nuclease. Importantly, p53 defects or depletion unexpectedly allow mutagenic RAD52 and POLθ pathways to hijack stalled forks, which we find reflected in p53 defective breast-cancer patient COSMIC mutational signatures. These data uncover p53 as a keystone regulator of replication homeostasis within a DNA restart network. Mechanistically, this has important implications for development of resistance in cancer therapy. Combined, these results define an unexpected role for p53-mediated suppression of replication genome instability.
传统上,p53肿瘤抑制因子在转录、凋亡和细胞周期停滞中发挥作用。然而,复制介导的基因组不稳定性是肿瘤发生的一个重要组成部分,p53突变会促进肿瘤进展和耐药性。通过描绘人类和小鼠功能分离的p53等位基因,我们发现p53缺失和功能获得(GOF)突变在驱动基因组不稳定的停滞或受损DNA复制叉重新启动方面存在缺陷,这在基因上与转录激活是可分离的。通过检测单细胞中的蛋白质-DNA叉相互作用,我们揭示了一种由p53-MLL3介导的MRE11 DNA复制重新启动核酸酶的招募。重要的是,p53缺陷或缺失意外地允许诱变的RAD52和POLθ途径劫持停滞的复制叉,我们发现这在p53缺陷的乳腺癌患者COSMIC突变特征中有所体现。这些数据揭示了p53是DNA重新启动网络中复制稳态的关键调节因子。从机制上讲,这对癌症治疗中耐药性的发展具有重要意义。综合来看,这些结果定义了p53介导的抑制复制基因组不稳定性的一个意想不到的作用。