Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Matsumoto, Japan.
Department of Neurology, Graduate School of Medical Sciences, Kumamoto University, 1-1-1 Honjo, Chuo-ku, Kumamoto-shi, Kumamoto, 860-8556, Japan.
Orphanet J Rare Dis. 2018 Jan 17;13(1):6. doi: 10.1186/s13023-017-0726-x.
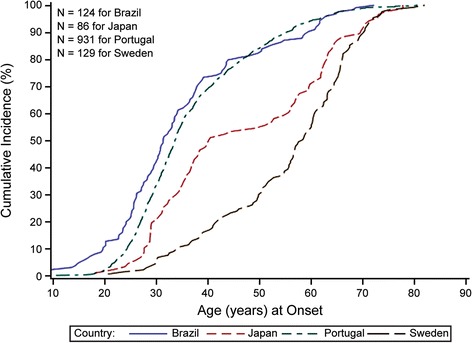
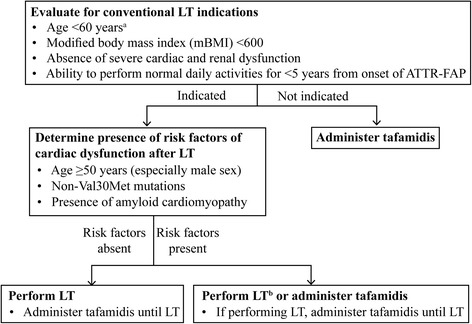
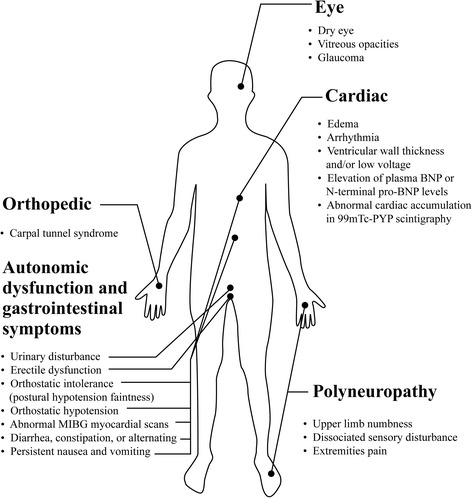
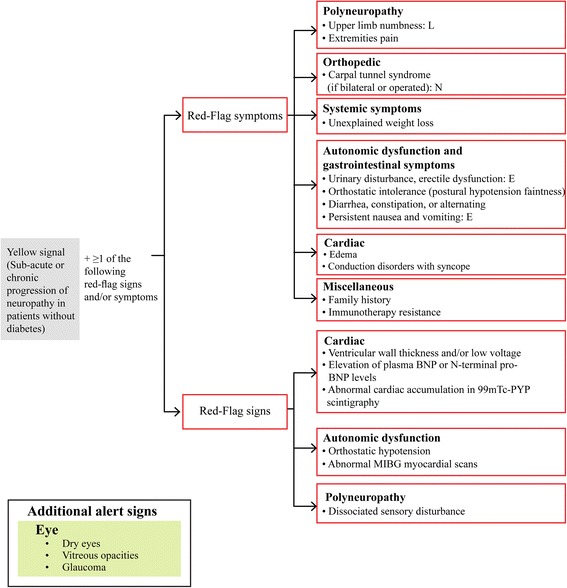
Hereditary ATTR (ATTRm) amyloidosis (also called transthyretin-type familial amyloid polyneuropathy [ATTR-FAP]) is an autosomal-dominant, adult-onset, rare systemic disorder predominantly characterized by irreversible, progressive, and persistent peripheral nerve damage. TTR gene mutations (e.g. replacement of valine with methionine at position 30 [Val30Met (p.Val50Met)]) lead to destabilization and dissociation of TTR tetramers into variant TTR monomers, which form amyloid fibrils that deposit in peripheral nerves and various organs, giving rise to peripheral and autonomic neuropathy and several non-disease specific symptoms.Phenotypic and genetic variability and non-disease-specific symptoms often delay diagnosis and lead to misdiagnosis. Red-flag symptom clusters simplify diagnosis globally. However, in Japan, types of TTR variants, age of onset, penetrance, and clinical symptoms of Val30Met are more varied than in other countries. Hence, development of a Japan-specific red-flag symptom cluster is warranted. Presence of progressive peripheral sensory-motor polyneuropathy and ≥1 red-flag sign/symptom (e.g. family history, autonomic dysfunction, cardiac involvement, carpal tunnel syndrome, gastrointestinal disturbances, unexplained weight loss, and immunotherapy resistance) suggests ATTR-FAP. Outside of Japan, pharmacotherapeutic options are first-line therapy. However, because of positive outcomes (better life expectancy and higher survival rates) with living donor transplant in Japan, liver transplantation remains first-line treatment, necessitating a Japan-specific treatment algorithm.Herein, we present a consolidated review of the ATTR-FAP Val30Met landscape in Japan and summarize findings from a medical advisory board meeting held in Tokyo on 18th August 2016, at which a Japan-specific ATTR-FAP red-flag symptom cluster and treatment algorithm was developed. Beside liver transplantation, a TTR-stabilizing agent (e.g. tafamidis) is a treatment option. Early diagnosis and timely treatment using the Japan-specific red-flag symptom cluster and treatment algorithm might help guide clinicians regarding apt and judicious use of available treatment modalities.
遗传性转甲状腺素蛋白淀粉样变性病(ATTRm)(也称为转甲状腺素蛋白型家族性淀粉样多发性神经病[ATTR-FAP])是一种常染色体显性遗传、成人发病、罕见的系统性疾病,主要表现为不可逆、进行性和持续性周围神经损伤。TTR 基因突变(例如缬氨酸被蛋氨酸取代,第 30 位[Val30Met(p.Val50Met)])导致 TTR 四聚体不稳定和解离为变异 TTR 单体,这些单体形成淀粉样纤维,沉积在周围神经和各种器官中,导致周围和自主神经病以及几种非疾病特异性症状。表型和遗传变异性以及非疾病特异性症状常导致诊断延迟和误诊。标志性症状群简化了全球诊断。然而,在日本,TTR 变异类型、发病年龄、外显率和 Val30Met 的临床症状比其他国家更为多样化。因此,有必要制定日本特有的标志性症状群。进行性周围感觉运动多发性神经病和≥1 个标志性体征/症状(例如家族史、自主神经功能障碍、心脏受累、腕管综合征、胃肠道紊乱、原因不明的体重减轻和免疫治疗抵抗)提示存在 ATTR-FAP。在日本以外,药物治疗选择是一线治疗。然而,由于在日本进行活体供肝移植可获得较好的预后(更长的预期寿命和更高的存活率),肝移植仍然是一线治疗,因此需要制定日本特有的治疗方案。本文总结了日本 ATTR-FAP Val30Met 疾病概况,并总结了 2016 年 8 月 18 日在东京举行的医学顾问委员会会议的结果,会上制定了日本特有的 ATTR-FAP 标志性症状群和治疗方案。除了肝移植,TTR 稳定剂(例如 tafamidis)也是一种治疗选择。使用日本特有的标志性症状群和治疗方案进行早期诊断和及时治疗,可能有助于指导临床医生合理使用现有治疗方法。