Henriques Sara F, Patissier Cécile, Bourg Nathalie, Fecchio Chiara, Sandona Doriana, Marsolier Justine, Richard Isabelle
INTEGRARE, Genethon, Inserm, Univ Evry, Université Paris-Saclay, Evry, France.
Department of Biomedical Sciences, University of Padova, Via U. Bassi, Padova, Italy.
PLoS One. 2018 Jan 23;13(1):e0191274. doi: 10.1371/journal.pone.0191274. eCollection 2018.
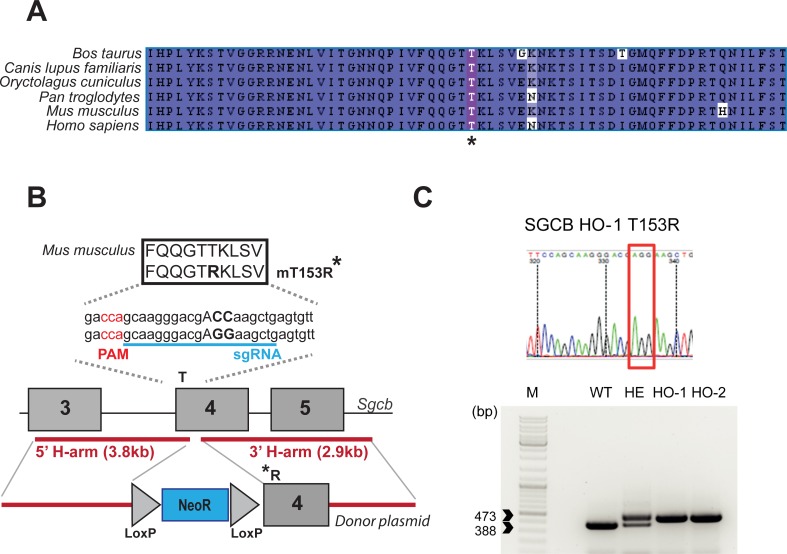
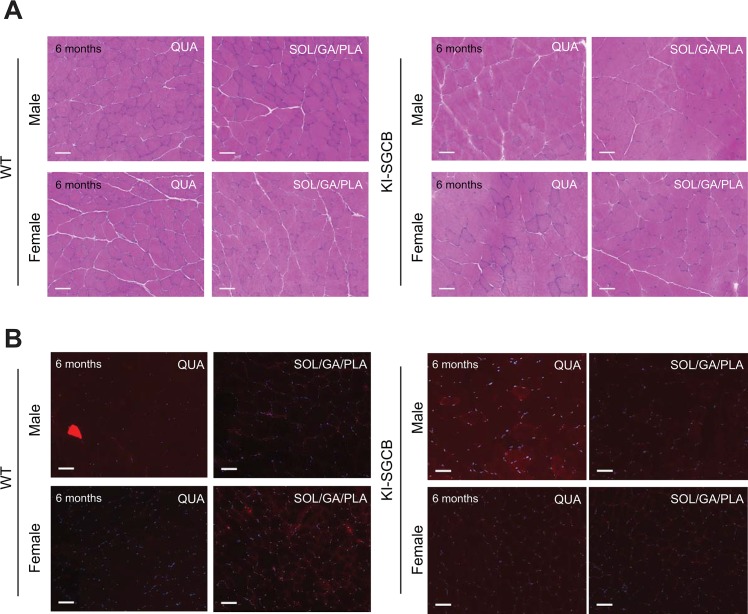
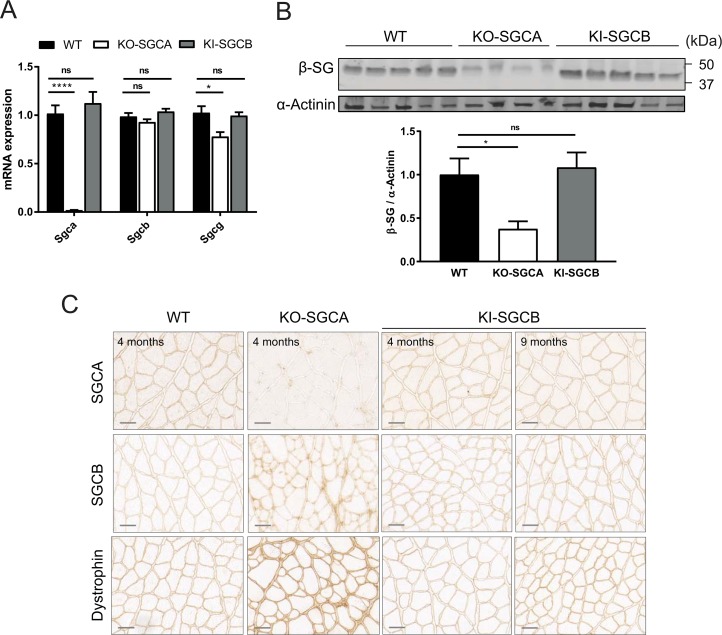
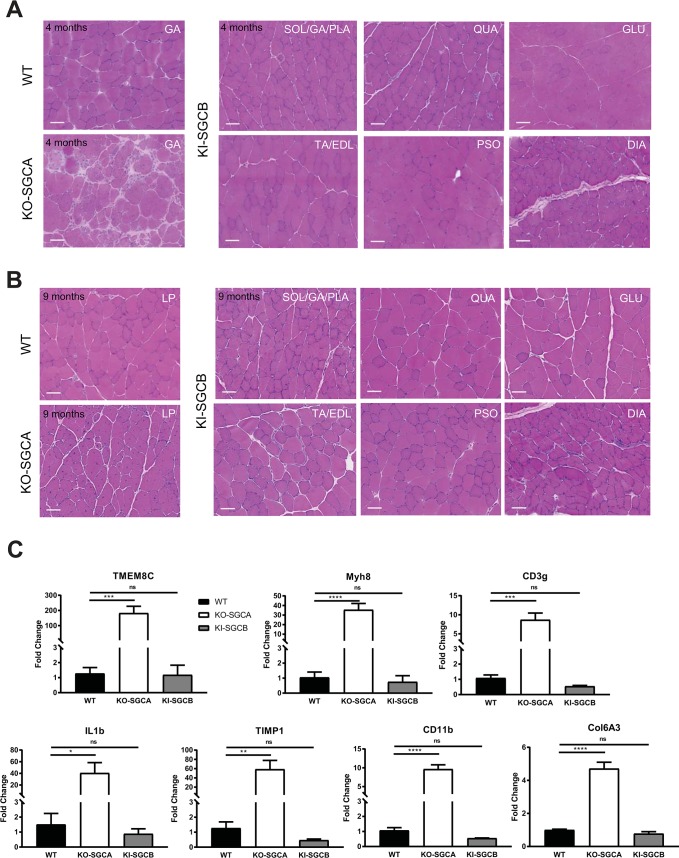
Sarcoglycanopathies are rare autosomic limb girdle muscular dystrophies caused by mutations in one of the genes coding for sarcoglycan (α, β, δ, and γ-sarcoglycans). Sarcoglycans form a complex, which is an important part of the dystrophin-associated glycoprotein complex that protects sarcolemma against muscle contraction-induced damages. Absence of one of the sarcoglycan at the plasma membrane induces the disappearance of the whole complex and perturbs muscle fiber membrane integrity. We previously demonstrated that point mutations in the human sarcoglycan genes affects the folding of the corresponding protein, which is then retained in the endoplasmic reticulum by the protein quality control and prematurely degraded by the proteasome. Interestingly, modulation of the quality control using pharmacological compounds allowed the rescue of the membrane localization of the mutated sarcoglycan. Two previously generated mouse models, knock-in for the most common sarcoglycan mutant, R77C α-sarcoglycan, failed in reproducing the dystrophic phenotype observed in human patients. Based on these results and the need to test therapies for these fatal diseases, we decided to generate a new knock-in mouse model carrying the missense mutation T151R in the β-sarcoglycan gene since this is the second sarcoglycan protein with the most frequently reported missense mutations. Muscle analysis, performed at the age of 4 and 9-months, showed the presence of the mutated β-sarcoglycan protein and of the other components of the dystrophin-associated glycoprotein complex at the muscle membrane. In addition, these mice did not develop a dystrophic phenotype, even at a late stage or in condition of stress-inducing exercise. We can speculate that the absence of phenotype in mouse may be due to a higher tolerance of the endoplasmic reticulum quality control for amino-acid changes in mice compared to human.
肌聚糖病是一种罕见的常染色体隐性肢带型肌营养不良症,由编码肌聚糖(α、β、δ和γ - 肌聚糖)的基因之一发生突变引起。肌聚糖形成一个复合体,它是抗肌萎缩蛋白相关糖蛋白复合体的重要组成部分,可保护肌膜免受肌肉收缩引起的损伤。质膜上缺少一种肌聚糖会导致整个复合体消失,并扰乱肌纤维膜的完整性。我们之前证明,人类肌聚糖基因中的点突变会影响相应蛋白质的折叠,该蛋白质随后会被蛋白质质量控制系统保留在内质网中,并被蛋白酶体过早降解。有趣的是,使用药物化合物调节质量控制可以挽救突变型肌聚糖的膜定位。之前生成的两种小鼠模型,即最常见的肌聚糖突变体R77C α - 肌聚糖的基因敲入模型,未能重现人类患者中观察到的营养不良表型。基于这些结果以及测试这些致命疾病治疗方法的需求,我们决定生成一种新的基因敲入小鼠模型,该模型在β - 肌聚糖基因中携带错义突变T151R,因为这是第二种错义突变报道频率最高的肌聚糖蛋白。在4个月和9个月大时进行的肌肉分析表明,在肌膜上存在突变的β - 肌聚糖蛋白以及抗肌萎缩蛋白相关糖蛋白复合体的其他成分。此外,这些小鼠即使在晚期或在应激诱导运动的情况下也没有出现营养不良表型。我们可以推测,小鼠中没有出现表型可能是因为与人类相比,内质网质量控制对小鼠氨基酸变化的耐受性更高。