Institute of Medical Genetics, University Hospital of Wales, Cardiff CF14 4XW, UK.
Division of Cancer and Genetics, School of Medicine, Cardiff University, Cardiff CF14 4XN, UK.
Brain. 2018 Mar 1;141(3):698-712. doi: 10.1093/brain/awx358.
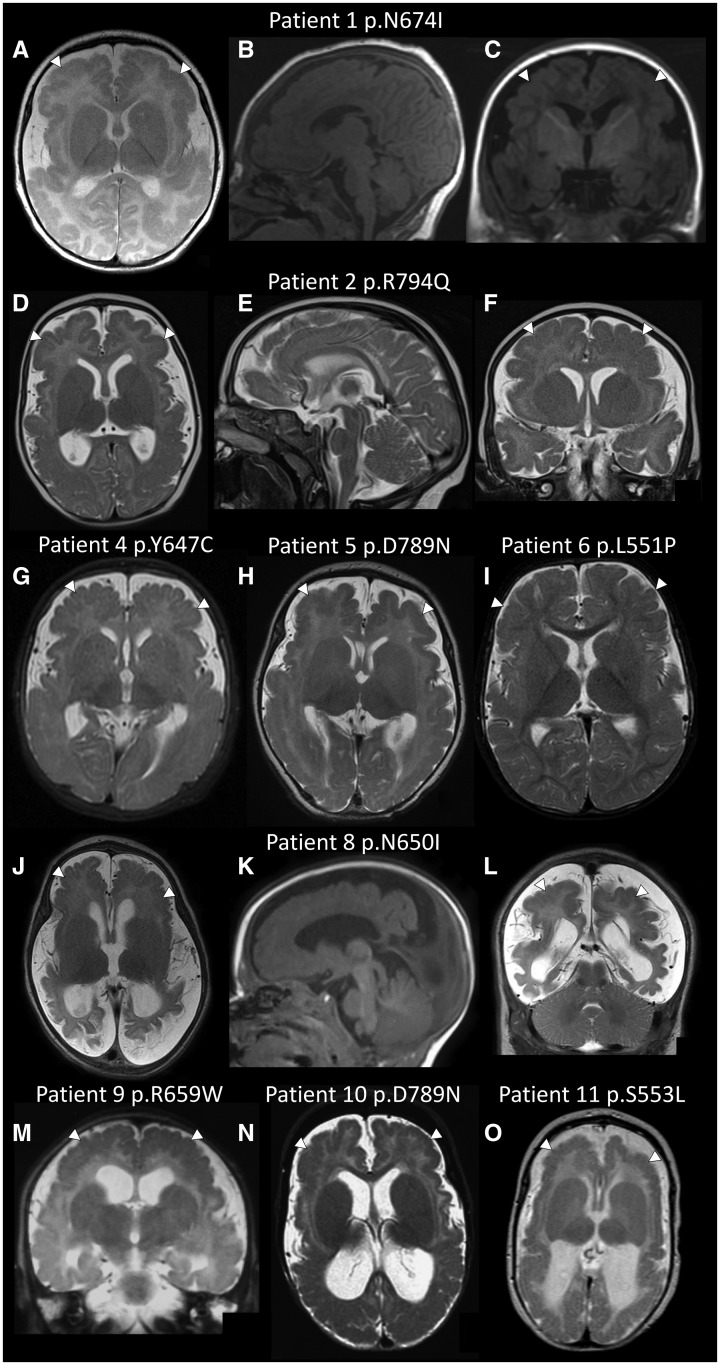
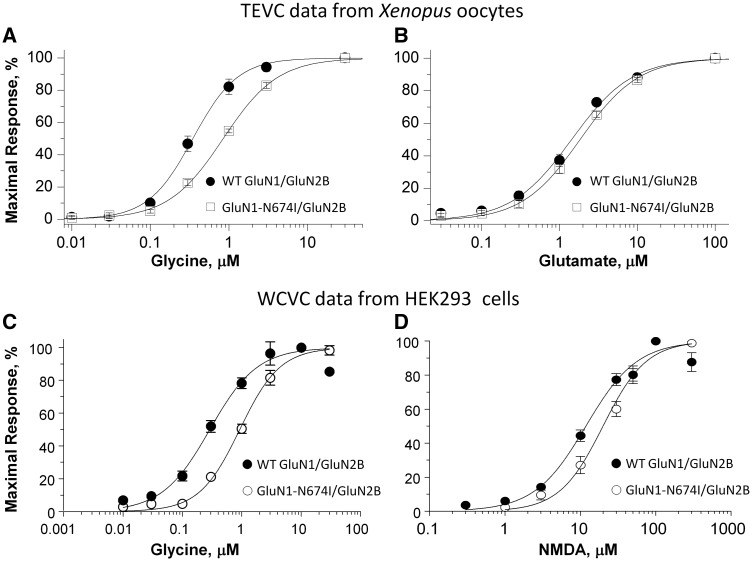
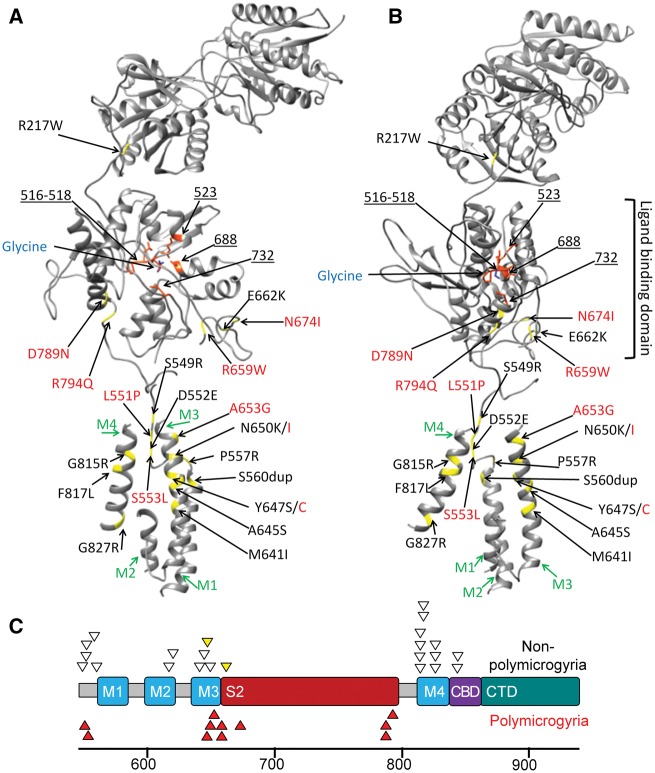
Polymicrogyria is a malformation of cortical development. The aetiology of polymicrogyria remains poorly understood. Using whole-exome sequencing we found de novo heterozygous missense GRIN1 mutations in 2 of 57 parent-offspring trios with polymicrogyria. We found nine further de novo missense GRIN1 mutations in additional cortical malformation patients. Shared features in the patients were extensive bilateral polymicrogyria associated with severe developmental delay, postnatal microcephaly, cortical visual impairment and intractable epilepsy. GRIN1 encodes GluN1, the essential subunit of the N-methyl-d-aspartate receptor. The polymicrogyria-associated GRIN1 mutations tended to cluster in the S2 region (part of the ligand-binding domain of GluN1) or the adjacent M3 helix. These regions are rarely mutated in the normal population or in GRIN1 patients without polymicrogyria. Using two-electrode and whole-cell voltage-clamp analysis, we showed that the polymicrogyria-associated GRIN1 mutations significantly alter the in vitro activity of the receptor. Three of the mutations increased agonist potency while one reduced proton inhibition of the receptor. These results are striking because previous GRIN1 mutations have generally caused loss of function, and because N-methyl-d-aspartate receptor agonists have been used for many years to generate animal models of polymicrogyria. Overall, our results expand the phenotypic spectrum associated with GRIN1 mutations and highlight the important role of N-methyl-d-aspartate receptor signalling in the pathogenesis of polymicrogyria.
脑回小畸形是一种皮质发育畸形。脑回小畸形的病因仍知之甚少。我们通过全外显子组测序,在 57 个有脑回小畸形的亲子三家中发现了 2 例 GRIN1 基因突变。我们在其他皮质畸形患者中发现了另外 9 例新发错义 GRIN1 突变。患者的共同特征是广泛的双侧脑回小畸形,伴有严重的发育迟缓、产后小头畸形、皮质视觉障碍和难治性癫痫。GRIN1 编码 GluN1,是 N-甲基-D-天冬氨酸受体的必需亚单位。与脑回小畸形相关的 GRIN1 突变倾向于聚集在 S2 区域(GluN1 配体结合域的一部分)或相邻的 M3 螺旋。这些区域在正常人群或没有脑回小畸形的 GRIN1 患者中很少发生突变。我们使用双电极和全细胞膜片钳分析表明,与脑回小畸形相关的 GRIN1 突变显著改变了受体的体外活性。其中 3 种突变增加了激动剂的效力,而 1 种突变降低了受体对质子的抑制。这些结果令人惊讶,因为以前的 GRIN1 突变通常导致功能丧失,而且 N-甲基-D-天冬氨酸受体激动剂已被用于多年来产生脑回小畸形的动物模型。总体而言,我们的研究结果扩展了与 GRIN1 突变相关的表型谱,并强调了 N-甲基-D-天冬氨酸受体信号在脑回小畸形发病机制中的重要作用。