Ming Tong Glenna So, Chan Kaai Tung, Chang Xiaoyong, Che Chi-Ming
State Key Laboratory of Synthetic Chemistry , Institute of Molecular Functional Materials , Department of Chemistry , The University of Hong Kong , Pokfulam Road , Hong Kong SAR , China . Email:
HKU Shenzhen Institute of Research and Innovation , Shenzhen 518053 , China . Email:
Chem Sci. 2015 May 1;6(5):3026-3037. doi: 10.1039/c4sc03697b. Epub 2015 Mar 10.
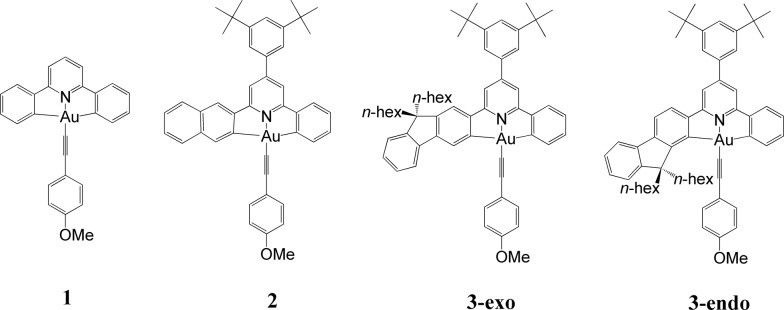
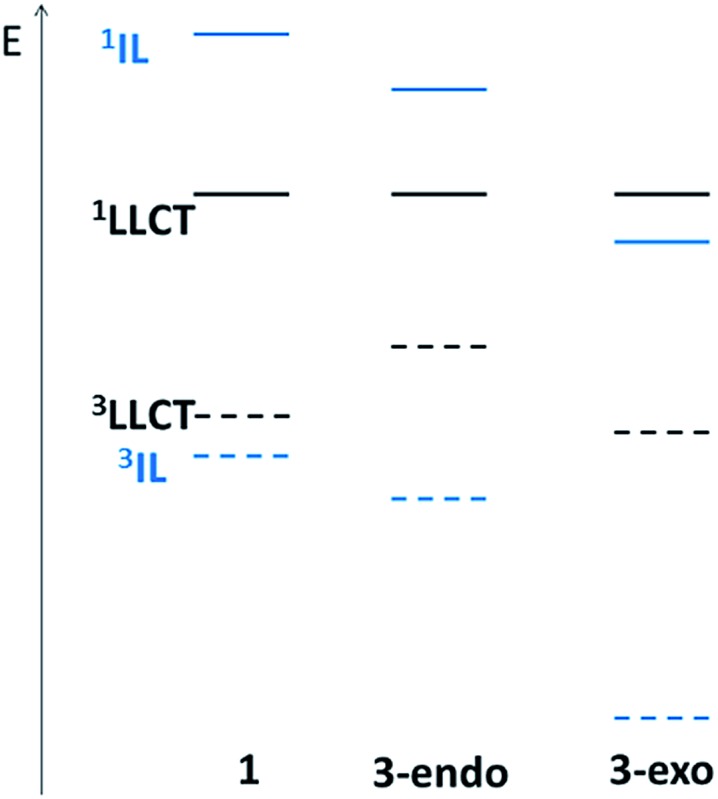
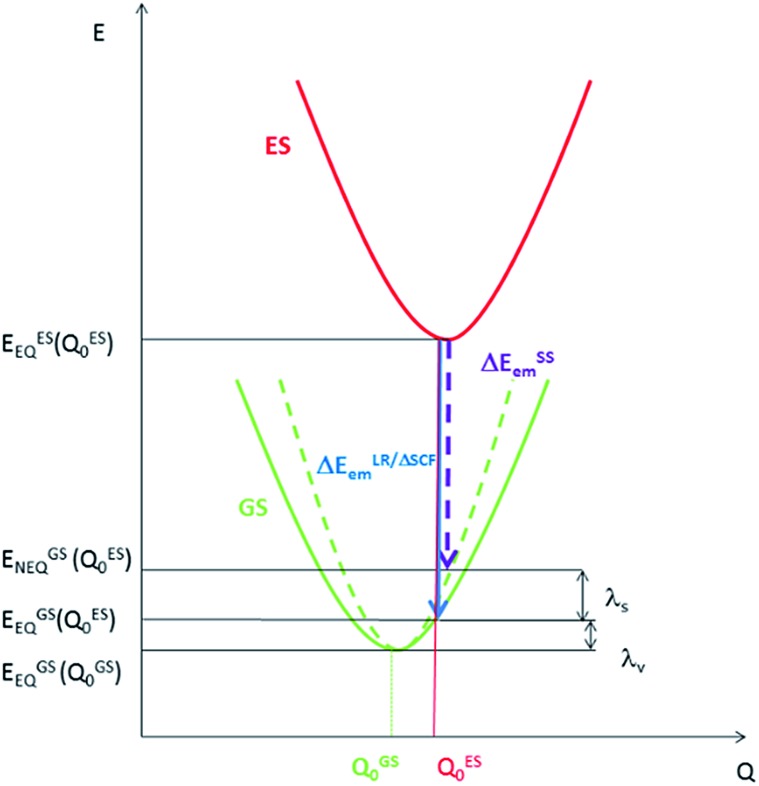
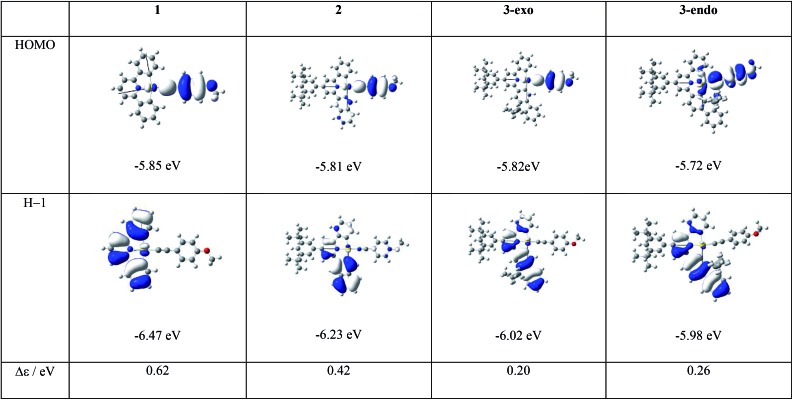
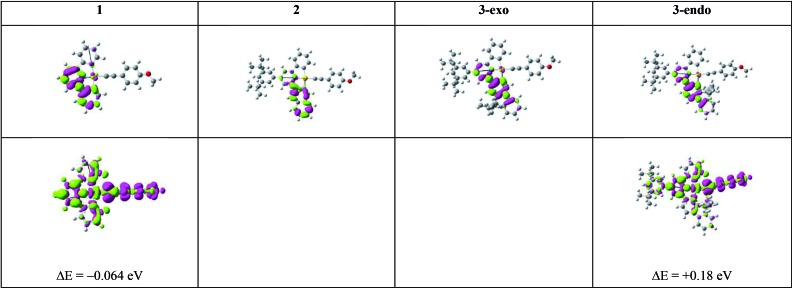
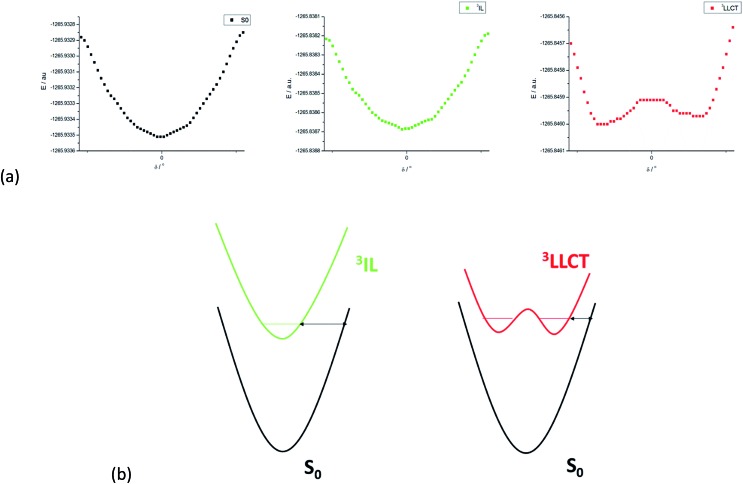
We have performed theoretical analyses of the photophysical properties of a series of cyclometalated gold(iii) arylacetylide complexes, [(C^N^C)AuC[triple bond, length as m-dash]CPh-4-OMe], with different extents of π-conjugation at the doubly C-deprotonated [C^N^C] ligand replacement of one of the phenyl moieties in the non-conjugated C^N^C ligand () by a naphthalenyl () or a fluorenyl moiety ( and ; HC^N^CH = 2,6-diphenylpyridine). Conforming to the conventional wisdom that extended π-conjugation imposes rigidity on the structure of the IL(ππ*(C^N^C)) excited state (IL = intraligand), the calculated Huang-Rhys factors for the IL → S transition follow the order: > > ∼ , which corroborates the experimental non-radiative decay rate constants, : ≫ > , but not . Density Functional Theory (DFT) calculations revealed that there is an additional triplet excited state minimum of LLCT character (LLCT = ligand-to-ligand charge transfer; [π(C[triple bond, length as m-dash]CPh-4-OMe) → π*(C^N^C)]) for complexes and . This LLCT excited state, possessing a large out-of-plane torsional motion between the planes of the C^N^C and arylacetylide ligands, has a double minimum anharmonic potential energy surface along this torsional coordinate which leads to enhanced Franck-Condon overlap between the LLCT excited state and the ground state. Together with the larger spin-orbit coupling (SOC) and solvent reorganization energy for the LLCT → S transition compared with those for the IL → S transition, the calculated values for the LLCT → S transition are more than 690- and 1500-fold greater than the corresponding IL → S transition for complexes and respectively. Importantly, when this LLCT → S decay channel is taken into consideration, the non-radiative decay rate constant could be reproduced and in the order of: ≫ , > . This challenges the common view that the facile non-radiative decay rate of transition metal complexes is due to the presence of a low-lying metal-centred dd or LMCT excited state (LMCT = ligand-to-metal charge transfer). By analysis of the relative order of MOs of the chromophoric [C^N^C] cyclometalated and arylacetylide ligands, one may discern why complexes and have a low-lying LLCT excited state while does not.
我们对一系列环金属化金(III)芳基乙炔配合物[(C^N^C)AuC≡CPh - 4 - OMe]的光物理性质进行了理论分析,这些配合物在双去质子化的[C^N^C]配体上具有不同程度的π共轭,即在非共轭C^N^C配体()中的一个苯基部分被萘基()或芴基部分(和;HC^N^CH = 2,6 - 二苯基吡啶)取代。符合传统观点,即扩展的π共轭使IL(ππ*(C^N^C))激发态(IL = 配体内)的结构具有刚性,计算得到的IL→S跃迁的黄 - 里斯因子遵循以下顺序:> > ∼,这与实验得到的非辐射衰变率常数相佐证,:≫ > ,但不包括。密度泛函理论(DFT)计算表明,对于配合物和,存在一个具有LLCT特征(LLCT = 配体到配体电荷转移;[π(C≡CPh - 4 - OMe)→π*(C^N^C)])的额外三重激发态最小值。这个LLCT激发态在C^N^C和芳基乙炔配体平面之间具有大的面外扭转运动,沿着这个扭转坐标具有双最小值非谐势能面,这导致LLCT激发态与基态之间的弗兰克 - 康登重叠增强。与IL→S跃迁相比,LLCT→S跃迁具有更大的自旋 - 轨道耦合(SOC)和溶剂重组能,计算得到的配合物和的LLCT→S跃迁的 值分别比相应的IL→S跃迁大690倍和1500倍以上。重要的是,当考虑这个LLCT→S衰变通道时,非辐射衰变率常数可以被重现,且顺序为:≫ ,> 。这挑战了常见观点,即过渡金属配合物容易发生非辐射衰变率是由于存在低能的以金属为中心的dd或LMCT激发态(LMCT = 配体到金属电荷转移)。通过分析发色团[C^N^C]环金属化和芳基乙炔配体的分子轨道的相对顺序,可以看出为什么配合物和具有低能的LLCT激发态而没有。