Torres-Rojas Francisco Israel, Alarcón-Romero Luz Del Carmen, Leyva-Vázquez Marco Antonio, Ortiz-Ortiz Julio, Mendoza-Catalán Miguel Ángel, Hernández-Sotelo Daniel, Del Moral-Hernández Oscar, Rodríguez-Ruiz Hugo Alberto, Leyva-Illades Dinorah, Flores-Alfaro Eugenia, Illades-Aguiar Berenice
Laboratory of Molecular Biomedicine, School of Chemical and Biological Sciences, Universidad Autónoma de Guerrero, Chilpancingo, Guerrero 39090, Mexico.
Laboratory of Cytopathology and Histochemistry, School of Chemical and Biological Sciences, Universidad Autónoma de Guerrero, Chilpancingo, Guerrero 39090, Mexico.
Oncol Lett. 2018 Feb;15(2):2278-2286. doi: 10.3892/ol.2017.7596. Epub 2017 Dec 13.
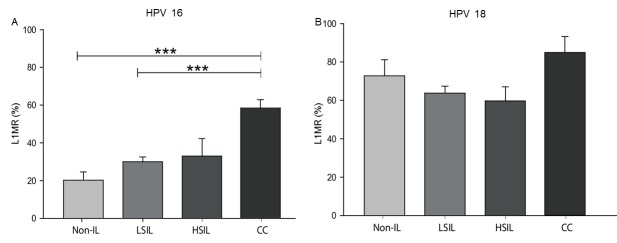
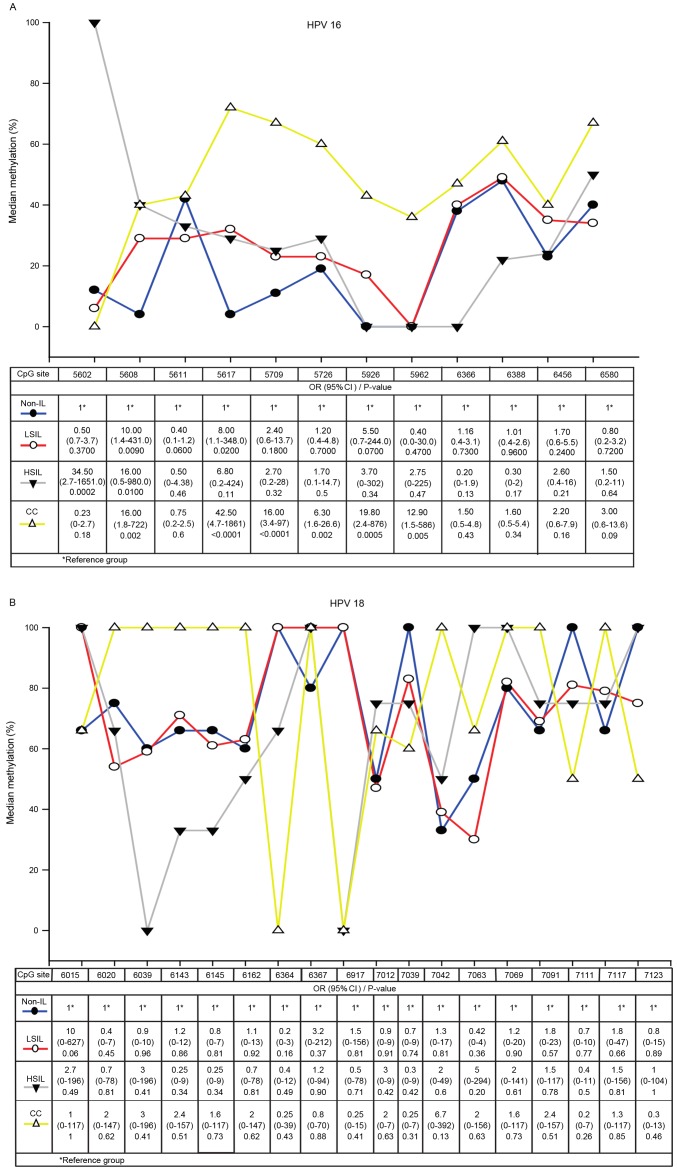
High-risk human papillomavirus (HPV) is the primary cause of cervical carcinoma (CC). Viral integration into the host chromosomes is associated with neoplastic progression, and epigenetic changes may occur as a result. The objective of the present study was to analyze HPV L1 gene methylation and to compare the use of quantitative polymerase chain reaction (qPCR), hybridization (ISH) and L1 methylation analysis as methods for detecting HPV integration. Cervical scrapes or biopsy samples positive for HPV 16 or 18, from 187 female patients with CC, squamous intraepithelial lesions (SILs) or no intraepithelial lesion (non-IL) were analyzed. Methylation of the L1 gene was determined using bisulfite modification followed by PCR, and HPV integration was subsequently analyzed. HPV 16 L1 gene methylation was revealed to increase with histological grade, with statistically significant differences observed as follows: Low-grade SIL vs. CC, P<0.0001 and non-IL vs. CC, P<0.0001. HPV 18 L1 gene methylation also increased according to histological grade, however, no statistically significant differences were observed. Methylation at CpG site 5608 of the HPV 16 L1 gene was associated with all grades of cervical lesions, whereas methylation at CpG site 5617 demonstrated the strongest association with CC (odds ratio, 42.5; 95% confidence interval, 4.7-1861; P<0.0001). The concordance rates between the various methods for the detection of the physical status of HPV 16 and HPV 18 were 96.1% for qPCR and ISH, 76.7% for qPCR and L1 gene methylation, and 84.8% for ISH and L1 gene methylation. In conclusion, methylation of the HPV 16 L1 gene increases significantly according to the grade of the cervical lesion, and methylation at CpG sites 5608 and 5617 of this gene may be used as prognostic biomarkers. ISH and L1 gene methylation have good concordance with qPCR with regards to the detection of HPV integration. Therefore, these are useful methods in determining the physical state of HPV.
高危型人乳头瘤病毒(HPV)是宫颈癌(CC)的主要病因。病毒整合入宿主染色体与肿瘤进展相关,可能会导致表观遗传学改变。本研究的目的是分析HPV L1基因甲基化,并比较定量聚合酶链反应(qPCR)、杂交(ISH)和L1甲基化分析作为检测HPV整合方法的应用情况。对187例患有CC、鳞状上皮内病变(SILs)或无上皮内病变(非IL)的女性患者的宫颈刮片或活检样本进行分析,这些样本HPV 16或18检测呈阳性。采用亚硫酸氢盐修饰后PCR的方法测定L1基因的甲基化,随后分析HPV整合情况。结果显示,HPV 16 L1基因甲基化随组织学分级增加,在以下比较中观察到具有统计学意义的差异:低级别SIL与CC相比,P<0.0001;非IL与CC相比,P<0.0001。HPV 18 L1基因甲基化也随组织学分级增加,但未观察到具有统计学意义的差异。HPV 16 L1基因CpG位点5608的甲基化与所有级别的宫颈病变相关,而CpG位点5617的甲基化与CC的相关性最强(优势比,42.5;95%置信区间,4.7 - 1861;P<0.0001)。检测HPV 16和HPV 18物理状态的各种方法之间的一致性率如下:qPCR和ISH为96.1%,qPCR和L1基因甲基化为76.7%,ISH和L1基因甲基化为84.8%。总之,HPV 16 L1基因甲基化随宫颈病变分级显著增加,该基因CpG位点5608和5617的甲基化可作为预后生物标志物。ISH和L1基因甲基化在检测HPV整合方面与qPCR具有良好的一致性。因此,这些是确定HPV物理状态的有用方法。