Rekdal Silje L, Anmarkrud Jarl Andreas, Johnsen Arild, Lifjeld Jan T
Natural History Museum University of Oslo Oslo Norway.
Ecol Evol. 2018 Jan 7;8(3):1680-1692. doi: 10.1002/ece3.3757. eCollection 2018 Feb.
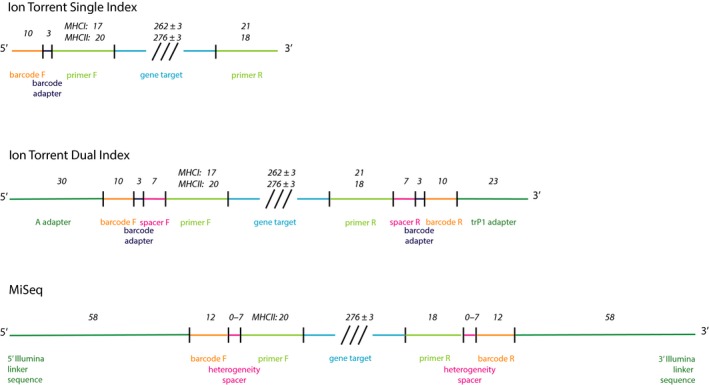
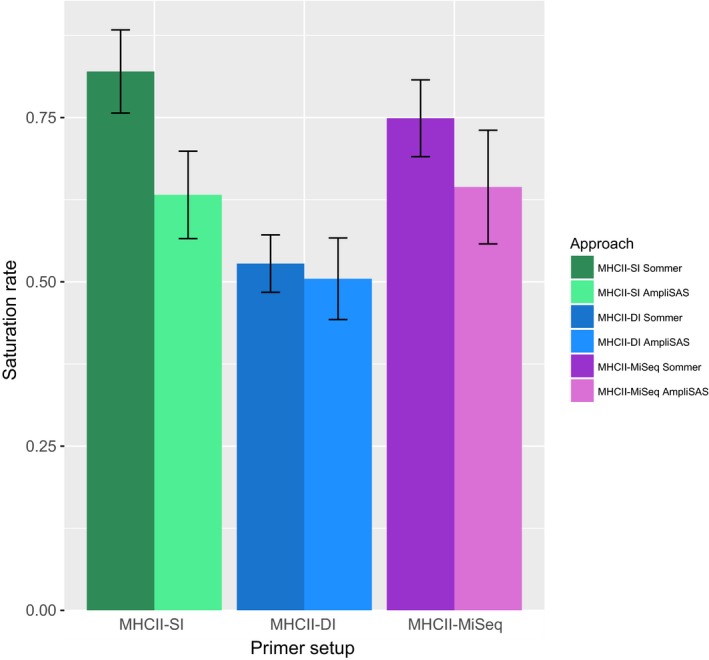
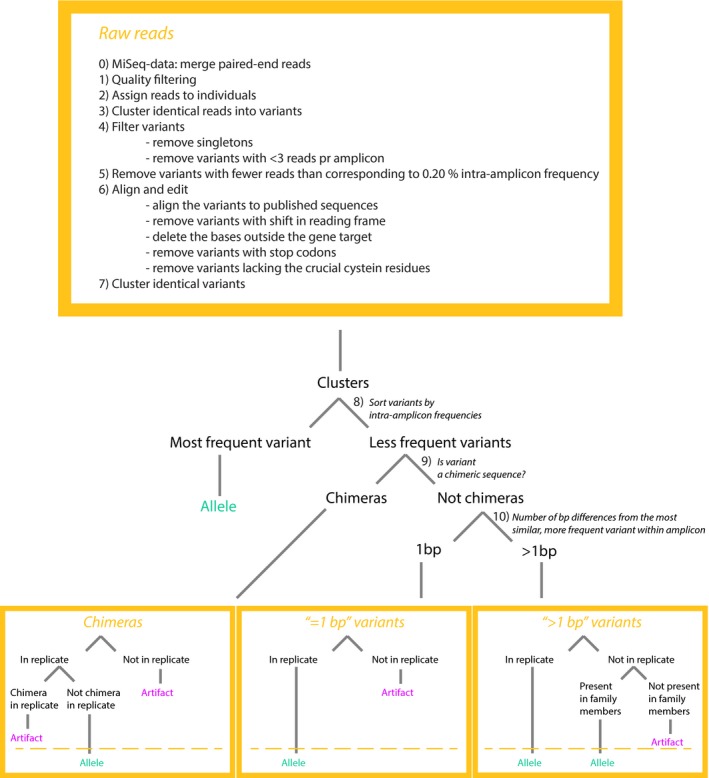
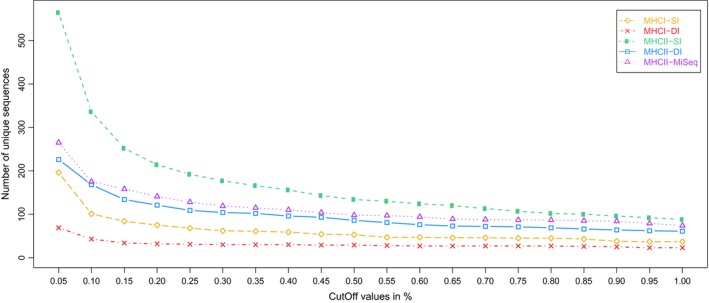
Genotyping of classical major histocompatibility complex (MHC) genes is challenging when they are hypervariable and occur in multiple copies. In this study, we used several different approaches to genotype the moderately variable MHC class I exon 3 (MHCIe3) and the highly polymorphic MHC class II exon 2 (MHCIIβe2) in the bluethroat (). Two family groups (eight individuals) were sequenced in replicates at both markers using Ion Torrent technology with both a single- and a dual-indexed primer structure. Additionally, MHCIIβe2 was sequenced on Illumina MiSeq. Allele calling was conducted by modifications of the pipeline developed by Sommer et al. (BMC Genomics, 14, 2013, 542) and the software AmpliSAS. While the different genotyping strategies gave largely consistent results for MHCIe3, with a maximum of eight alleles per individual, MHCIIβe2 was remarkably complex with a maximum of 56 MHCIIβe2 alleles called for one individual. Each genotyping strategy detected on average 50%-82% of all MHCIIβe2 alleles per individual, but dropouts were largely allele-specific and consistent within families for each strategy. The discrepancies among approaches indicate PCR biases caused by the platform-specific primer tails. Further, AmpliSAS called fewer alleles than the modified Sommer pipeline. Our results demonstrate that allelic dropout is a significant problem when genotyping the hypervariable MHCIIβe2. As these genotyping errors are largely nonrandom and method-specific, we caution against comparing genotypes across different genotyping strategies. Nevertheless, we conclude that high-throughput approaches provide a major advance in the challenging task of genotyping hypervariable MHC loci, even though they may not reveal the complete allelic repertoire.
对经典主要组织相容性复合体(MHC)基因进行基因分型具有挑战性,因为它们具有高度变异性且以多个拷贝形式存在。在本研究中,我们采用了几种不同的方法对蓝喉歌鸲中中度可变的MHC I类外显子3(MHCIe3)和高度多态的MHC II类外显子2(MHCIIβe2)进行基因分型。使用Ion Torrent技术,采用单索引和双索引引物结构,对两个家系组(8个个体)的两个标记进行了重复测序。此外,还在Illumina MiSeq上对MHCIIβe2进行了测序。通过对Sommer等人(《BMC基因组学》,2013年,第14卷,第542页)开发的流程和软件AmpliSAS进行修改来进行等位基因分型。虽然不同的基因分型策略对MHCIe3产生的结果基本一致,每个个体最多有8个等位基因,但MHCIIβe2却非常复杂,一个个体最多检测到56个MHCIIβe2等位基因。每种基因分型策略平均检测到每个个体所有MHCIIβe2等位基因的50%-82%,但缺失现象在很大程度上是等位基因特异性的,并且在每个家系内每种策略都是一致的。不同方法之间的差异表明平台特异性引物尾导致了PCR偏差。此外,AmpliSAS检测到的等位基因比修改后的Sommer流程少。我们的结果表明,对等位基因高度可变的MHCIIβe2进行基因分型时,等位基因缺失是一个重大问题。由于这些基因分型错误在很大程度上是非随机的且具有方法特异性,我们提醒不要在不同的基因分型策略之间比较基因型。尽管如此,我们得出结论,高通量方法在对高度可变的MHC位点进行基因分型这一具有挑战性的任务中取得了重大进展,即使它们可能无法揭示完整的等位基因库。