Razali Haslina, O'Connor Emily, Drews Anna, Burke Terry, Westerdahl Helena
Department of Animal and Plant Sciences, University of Sheffield, Sheffield, S10 2TN, UK.
Molecular Ecology and Evolution Lab, Department of Biology, Lund University, Sölvegatan 37, 223 62, Lund, Sweden.
BMC Res Notes. 2017 Jul 28;10(1):346. doi: 10.1186/s13104-017-2654-1.
High-throughput sequencing enables high-resolution genotyping of extremely duplicated genes. 454 amplicon sequencing (454) has become the standard technique for genotyping the major histocompatibility complex (MHC) genes in non-model organisms. However, illumina MiSeq amplicon sequencing (MiSeq), which offers a much higher read depth, is now superseding 454. The aim of this study was to quantitatively and qualitatively evaluate the performance of MiSeq in relation to 454 for genotyping MHC class I alleles using a house sparrow (Passer domesticus) dataset with pedigree information. House sparrows provide a good study system for this comparison as their MHC class I genes have been studied previously and, consequently, we had prior expectations concerning the number of alleles per individual.
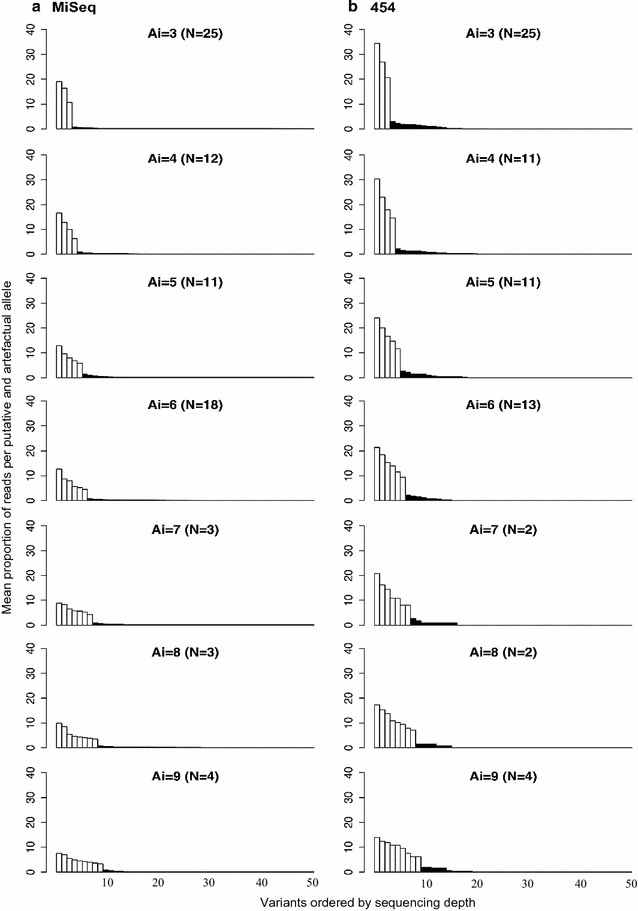
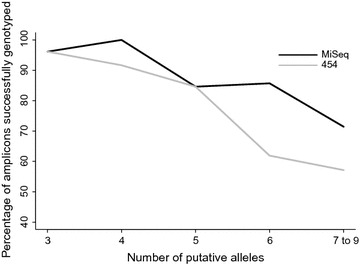
We found that 454 and MiSeq performed equally well in genotyping amplicons with low diversity, i.e. amplicons from individuals that had fewer than 6 alleles. Although there was a higher rate of failure in the 454 dataset in resolving amplicons with higher diversity (6-9 alleles), the same genotypes were identified by both 454 and MiSeq in 98% of cases.
We conclude that low diversity amplicons are equally well genotyped using either 454 or MiSeq, but the higher coverage afforded by MiSeq can lead to this approach outperforming 454 in amplicons with higher diversity.
高通量测序能够对高度重复的基因进行高分辨率基因分型。454 扩增子测序(454)已成为对非模式生物中的主要组织相容性复合体(MHC)基因进行基因分型的标准技术。然而,具有更高读取深度的 Illumina MiSeq 扩增子测序(MiSeq)如今正在取代 454。本研究的目的是使用具有系谱信息的家麻雀(Passer domesticus)数据集,定量和定性地评估 MiSeq 相对于 454 在 MHC I 类等位基因基因分型方面的性能。家麻雀为这种比较提供了一个良好的研究系统,因为它们的 MHC I 类基因先前已被研究过,因此我们对每个个体的等位基因数量有先验预期。
我们发现,在对低多样性扩增子(即来自等位基因少于 6 个的个体的扩增子)进行基因分型时,454 和 MiSeq 的表现同样出色。尽管在 454 数据集中解析具有更高多样性(6 - 9 个等位基因)的扩增子时失败率更高,但在 98%的情况下,454 和 MiSeq 识别出的基因型相同。
我们得出结论,使用 454 或 MiSeq 对低多样性扩增子进行基因分型的效果同样良好,但 MiSeq 提供的更高覆盖率可能导致该方法在具有更高多样性的扩增子中表现优于 454。