Boido Marina, De Amicis Elena, Valsecchi Valeria, Trevisan Marco, Ala Ugo, Ruegg Markus A, Hettwer Stefan, Vercelli Alessandro
Department of Neuroscience Rita Levi Montalcini, Neuroscience Institute Cavalieri Ottolenghi, University of Turin, Turin, Italy.
Department of Molecular Biotechnology and Health Sciences, University of Turin, Turin, Italy.
Front Cell Neurosci. 2018 Jan 30;12:17. doi: 10.3389/fncel.2018.00017. eCollection 2018.
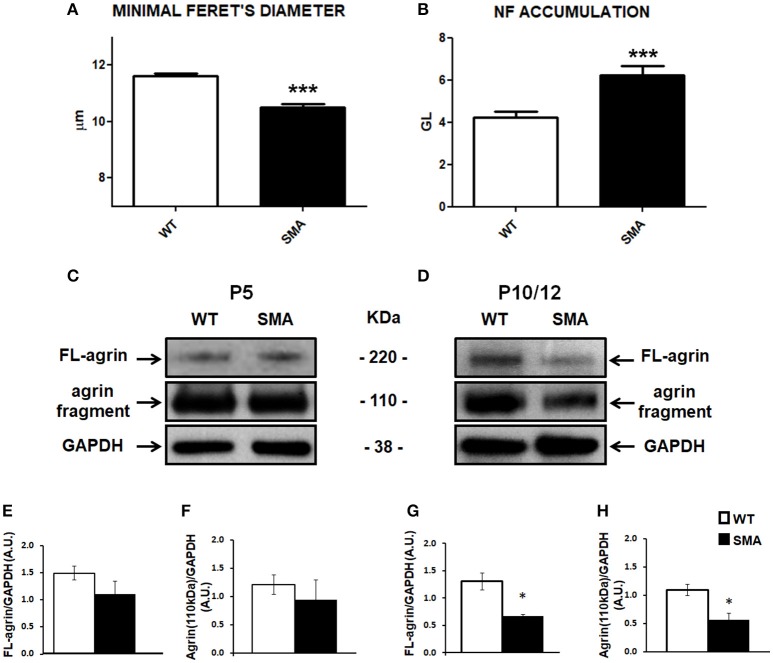
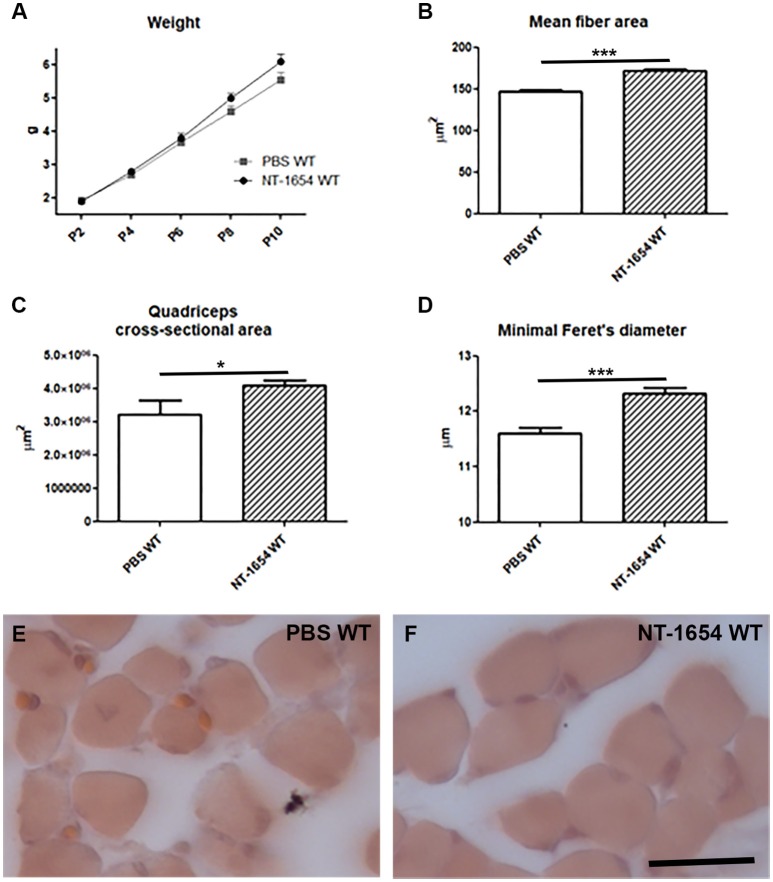
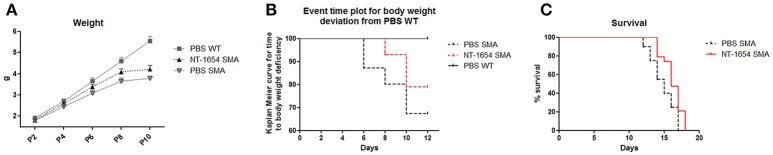
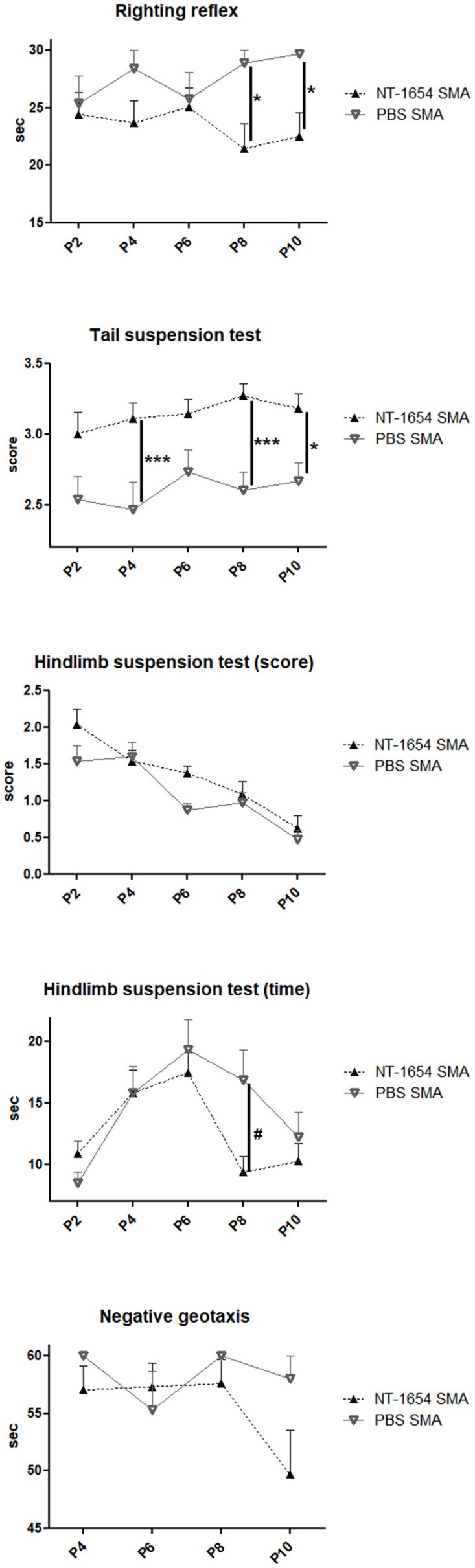
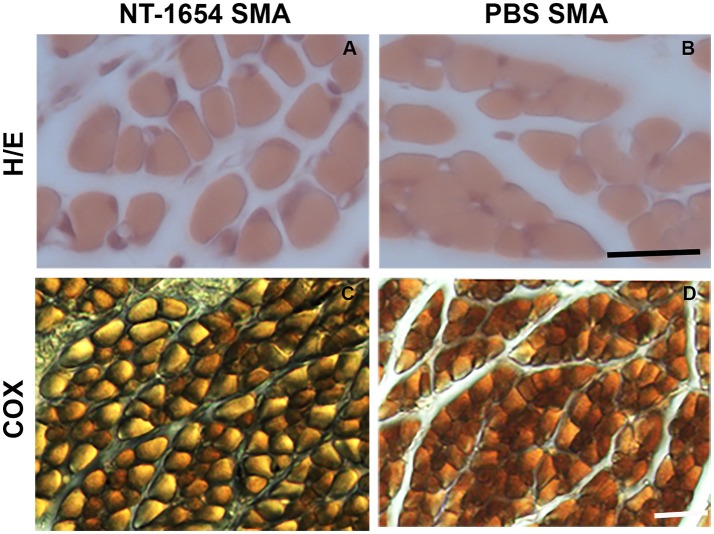
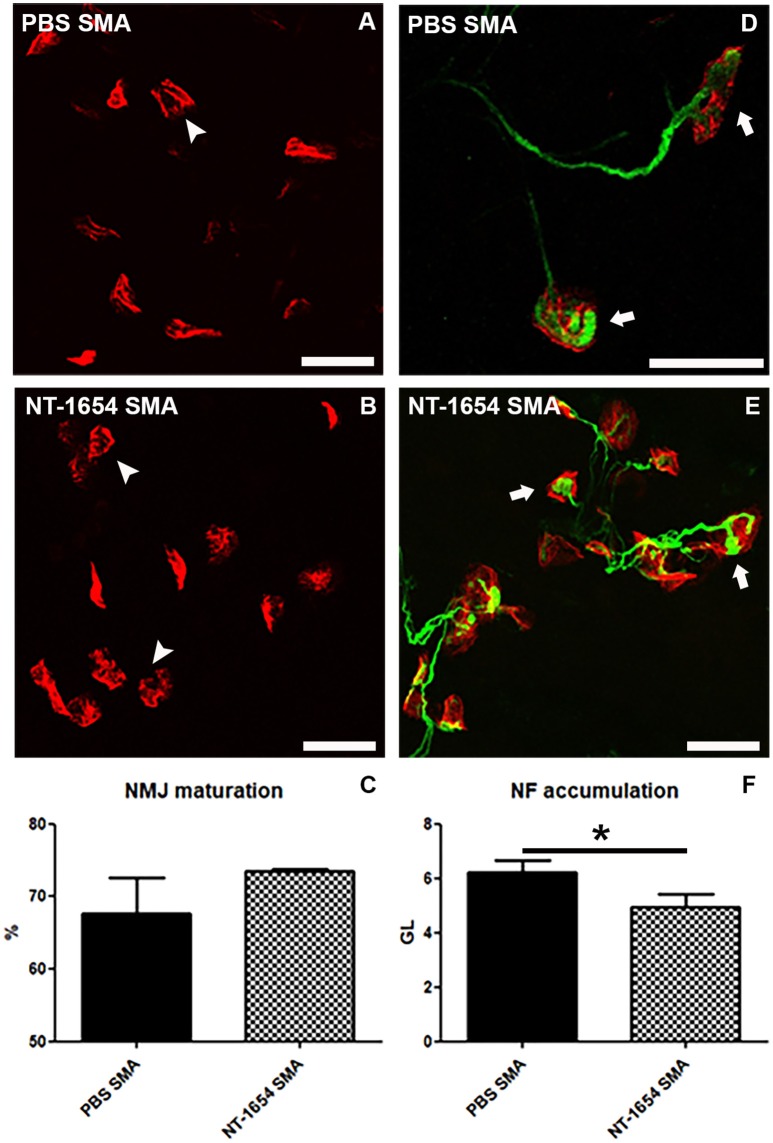
Spinal muscular atrophy (SMA) is a pediatric genetic disease, characterized by motor neuron (MN) death, leading to progressive muscle weakness, respiratory failure, and, in the most severe cases, to death. Abnormalities at the neuromuscular junction (NMJ) have been reported in SMA, including neurofilament (NF) accumulation at presynaptic terminals, immature and smaller than normal endplates, reduced transmitter release, and, finally, muscle denervation. Here we have studied the role of agrin in SMAΔ7 mice, the experimental model of SMAII. We observed a 50% reduction in agrin expression levels in quadriceps of P10 SMA mice compared to age-matched WT controls. To counteract such condition, we treated SMA mice from birth onwards with therapeutic agrin biological NT-1654, an active splice variant of agrin retaining synaptogenic properties, which is also resistant to proteolytic cleavage by neurotrypsin. Mice were analyzed for behavior, muscle and NMJ histology, and survival. Motor behavior was significantly improved and survival was extended by treatment of SMA mice with NT-1654. At P10, H/E-stained sections of the quadriceps, a proximal muscle early involved in SMA, showed that NT-1654 treatment strongly prevented the size decrease of muscle fibers. Studies of NMJ morphology on whole-mount diaphragm preparations revealed that NT-1654-treated SMA mice had more mature NMJs and reduced NF accumulation, compared to vehicle-treated SMA mice. We conclude that increasing agrin function in SMA has beneficial outcomes on muscle fibers and NMJs as the agrin biological NT-1654 restores the crosstalk between muscle and MNs, delaying muscular atrophy, improving motor performance and extending survival.
脊髓性肌萎缩症(SMA)是一种儿科遗传性疾病,其特征是运动神经元(MN)死亡,导致进行性肌肉无力、呼吸衰竭,在最严重的情况下会导致死亡。SMA中已报道神经肌肉接头(NMJ)存在异常,包括突触前末端神经丝(NF)积累、终板不成熟且比正常终板小、递质释放减少,最终导致肌肉失神经支配。在此,我们研究了聚集蛋白在SMAII实验模型SMAΔ7小鼠中的作用。我们观察到,与年龄匹配的野生型对照相比,P10 SMA小鼠股四头肌中聚集蛋白表达水平降低了50%。为了抵消这种情况,我们从出生起就用治疗性聚集蛋白生物制剂NT-1654治疗SMA小鼠,NT-1654是聚集蛋白的一种活性剪接变体,保留了突触生成特性,并且对神经胰蛋白酶的蛋白水解切割具有抗性。对小鼠的行为、肌肉和NMJ组织学以及存活率进行了分析。用NT-1654治疗SMA小鼠可显著改善运动行为并延长生存期。在P10时,对SMA早期累及的近端肌肉股四头肌进行苏木精/伊红(H/E)染色切片显示,NT-1654治疗强烈阻止了肌纤维尺寸减小。对整装膈膜制剂的NMJ形态学研究表明,与用赋形剂处理的SMA小鼠相比,用NT-1654治疗的SMA小鼠具有更成熟的NMJ且NF积累减少。我们得出结论,增加SMA中聚集蛋白的功能对肌纤维和NMJ具有有益效果,因为聚集蛋白生物制剂NT-1654恢复了肌肉和MN之间的相互作用,延缓了肌肉萎缩,改善了运动性能并延长了生存期。