Molecular Biology Center; State Key Laboratory of Trauma, Burn, and Combined Injury; Research Institute of Surgery and Daping Hospital, The Third Military Medical University, 10 Changjiang Zhilu, Chongqing, 400042, China.
Department of Neurology and Pharmacology, Boston University School of Medicine, Boston, MA, 02118, USA.
Cell Death Dis. 2018 Feb 14;9(2):252. doi: 10.1038/s41419-018-0316-4.
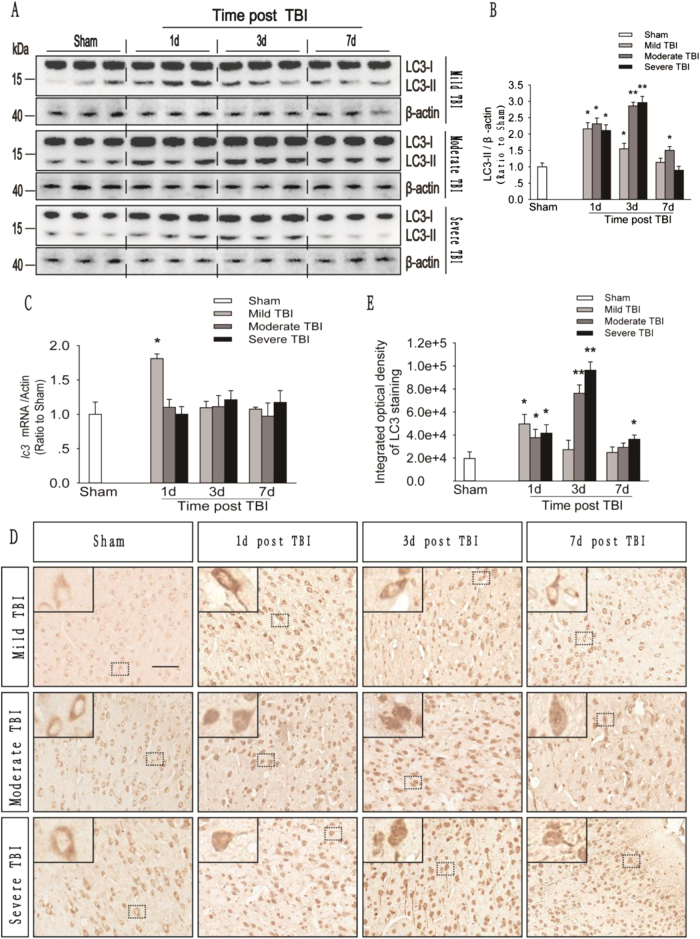
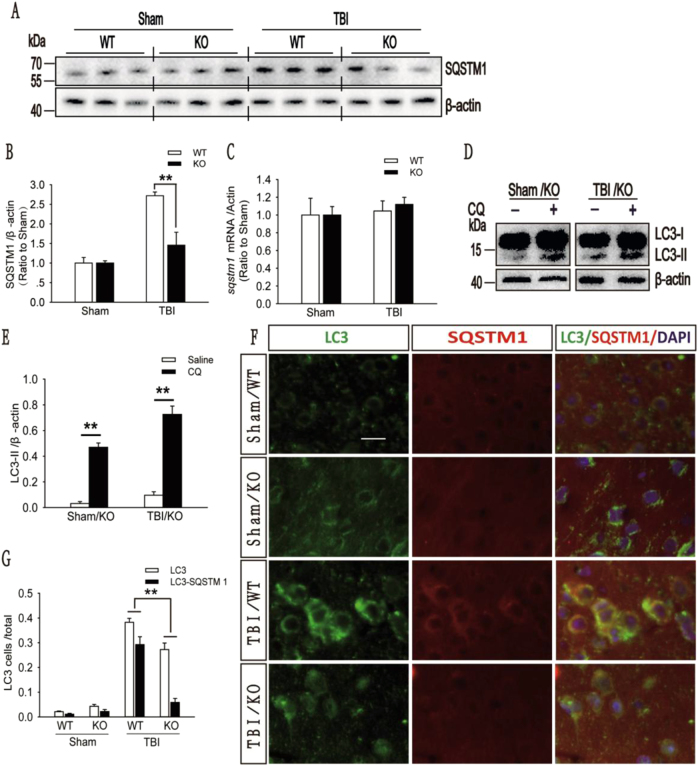
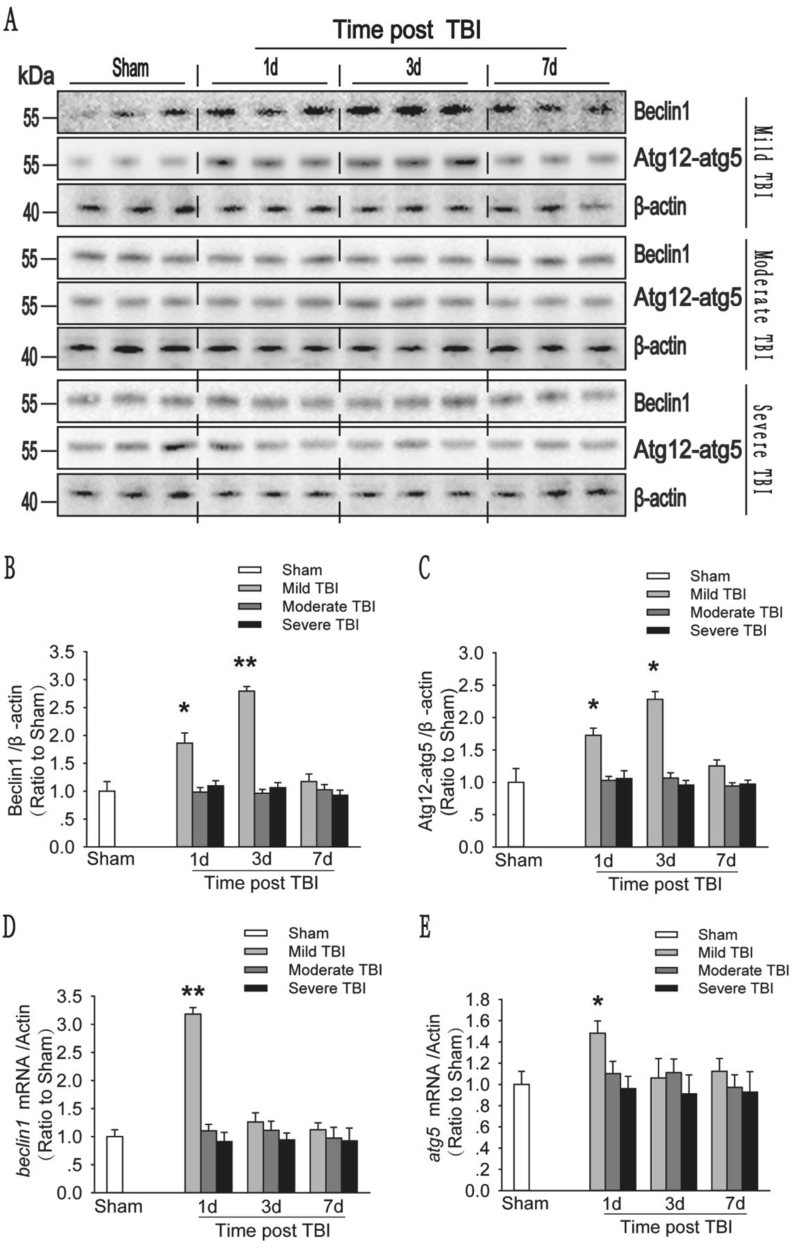
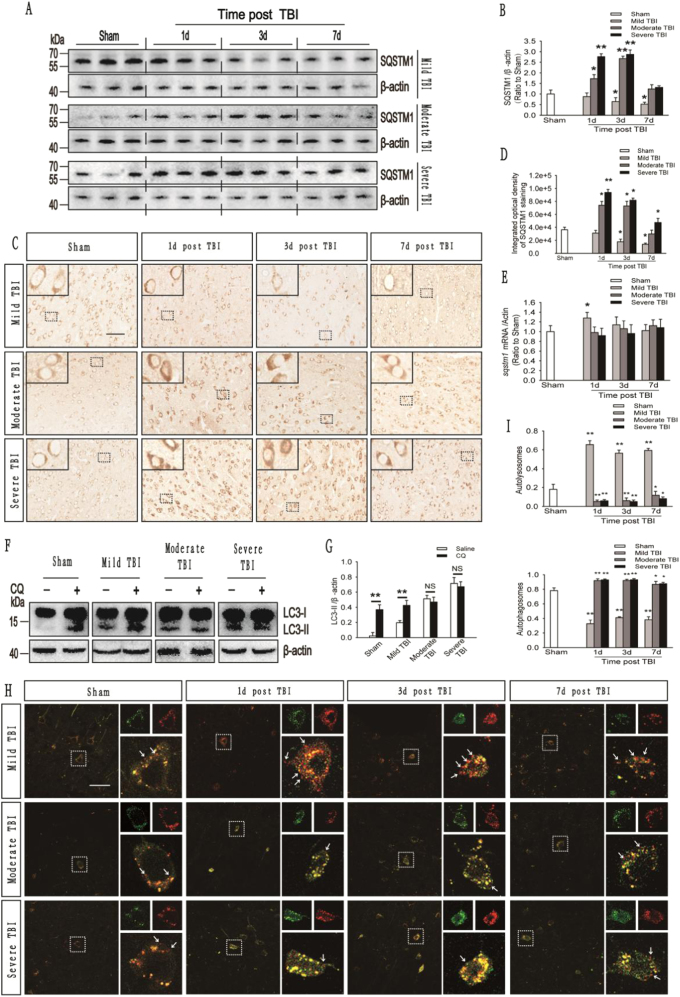
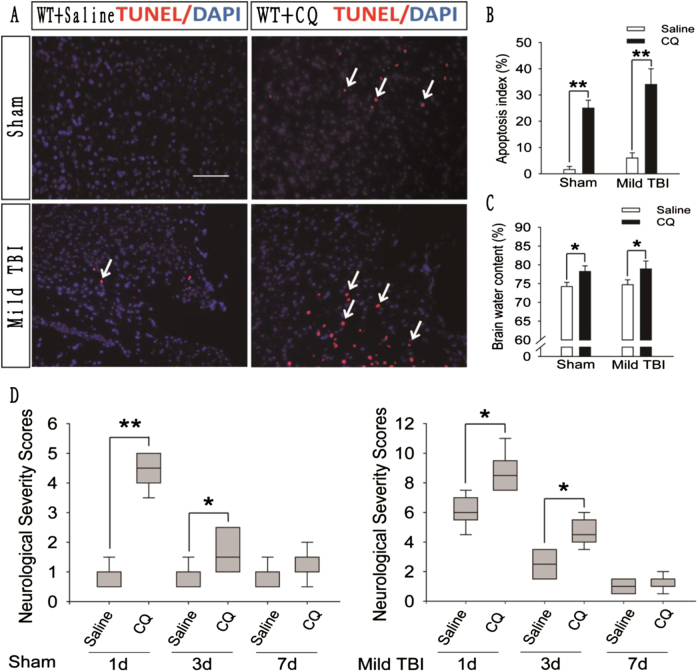
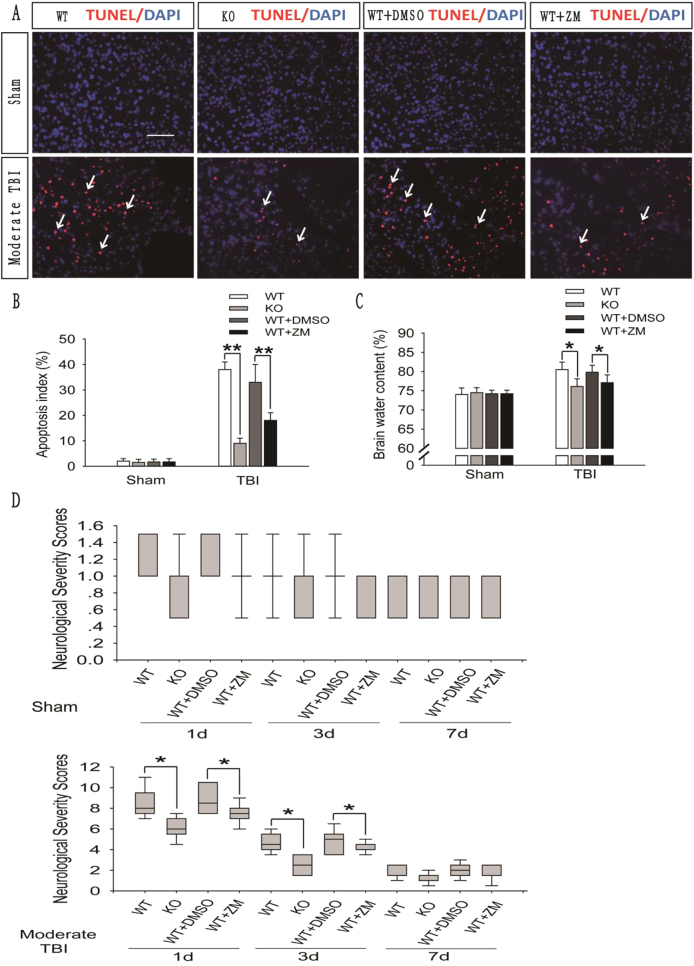
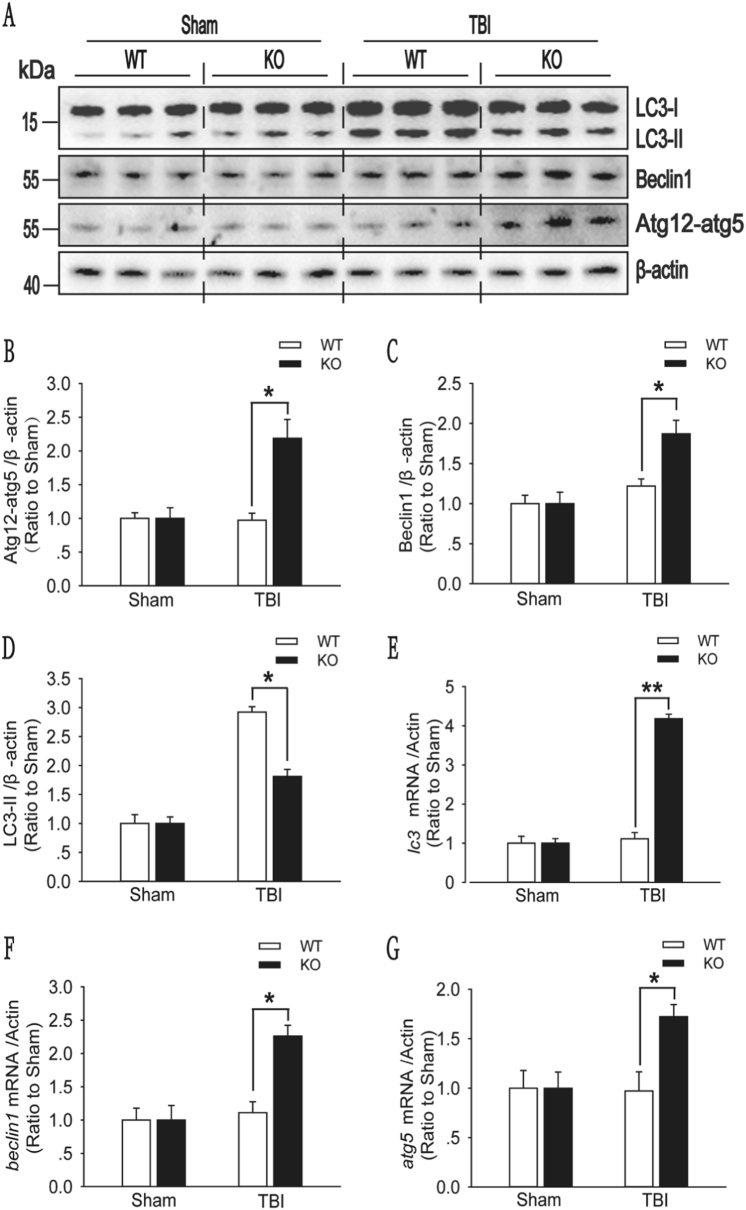
Recent studies have shown that after traumatic brain injury (TBI), the number of autophagosomes is markedly increased in brain cells surrounding the wound; however, whether autophagy is enhanced or suppressed by TBI remains controversial. In our study, we used a controlled cortical impact system to establish models of mild, moderate and severe TBI. In the mild TBI model, the levels of autophagy-related protein 6 (Beclin1) and autophagy-related protein 12 (ATG12)-autophagy-related protein 5 (ATG5) conjugates were increased, indicating the enhanced initiation of autophagy. Furthermore, the level of the autophagic substrate sequestosome 1 (SQSTM1) was decreased in the ipsilateral cortex. This result, together with the results observed in tandem mRFP-GFP-LC3 adeno-associated virus (AAV)-infected mice, indicates that autophagosome clearance was also increased after mild TBI. Conversely, following moderate and severe TBI, there was no change in the initiation of autophagy, and autophagosome accumulation was observed. Next, we used chloroquine (CQ) to artificially impair autophagic flux in the injured cortex of the mild TBI model and found that the severity of trauma was obviously exacerbated. In addition, autophagic flux and trauma severity were significantly improved in adenosine A receptor (AR) knockout (KO) mice subjected to moderate TBI. Thus, AR may be involved in regulating the impairment of autophagic flux in response to brain injury. Our findings suggest that whether autophagy is increased after TBI is associated with whether autophagic flux is impaired, and the impairment of autophagic flux exacerbates the severity of trauma. Furthermore, AR may be a target for alleviating the impairment in autophagic flux after TBI.
最近的研究表明,在创伤性脑损伤(TBI)后,伤口周围的脑细胞中自噬体的数量明显增加;然而,TBI 是否增强或抑制自噬仍存在争议。在我们的研究中,我们使用了皮质撞击控制系统来建立轻度、中度和重度 TBI 模型。在轻度 TBI 模型中,自噬相关蛋白 6(Beclin1)和自噬相关蛋白 12(ATG12)-自噬相关蛋白 5(ATG5)缀合物的水平增加,表明自噬的起始增强。此外,同侧皮质中自噬底物 sequestosome 1(SQSTM1)的水平降低。这一结果,以及串联 mRFP-GFP-LC3 腺相关病毒(AAV)感染小鼠的观察结果,表明轻度 TBI 后自噬体的清除也增加了。相反,中度和重度 TBI 后,自噬的起始没有变化,并且观察到自噬体的积累。接下来,我们使用氯喹(CQ)在轻度 TBI 模型损伤皮质中人为损害自噬流,发现创伤的严重程度明显加重。此外,在中度 TBI 的腺苷 A 受体(AR)敲除(KO)小鼠中,自噬流和创伤严重程度显著改善。因此,AR 可能参与调节脑损伤后自噬流的损害。我们的研究结果表明,TBI 后自噬是否增加与自噬流是否受损有关,自噬流的损害会加重创伤的严重程度。此外,AR 可能是减轻 TBI 后自噬流损害的靶点。