Institute of Physiology, University of Regensburg, Universitätsstraße 31, 93053, Regensburg, Germany.
MVZ Dialyse Alter Teichweg, Alter Teichweg 59-61, 22049, Hamburg, Germany.
Pflugers Arch. 2018 Jul;470(7):1127-1137. doi: 10.1007/s00424-018-2118-z. Epub 2018 Feb 17.
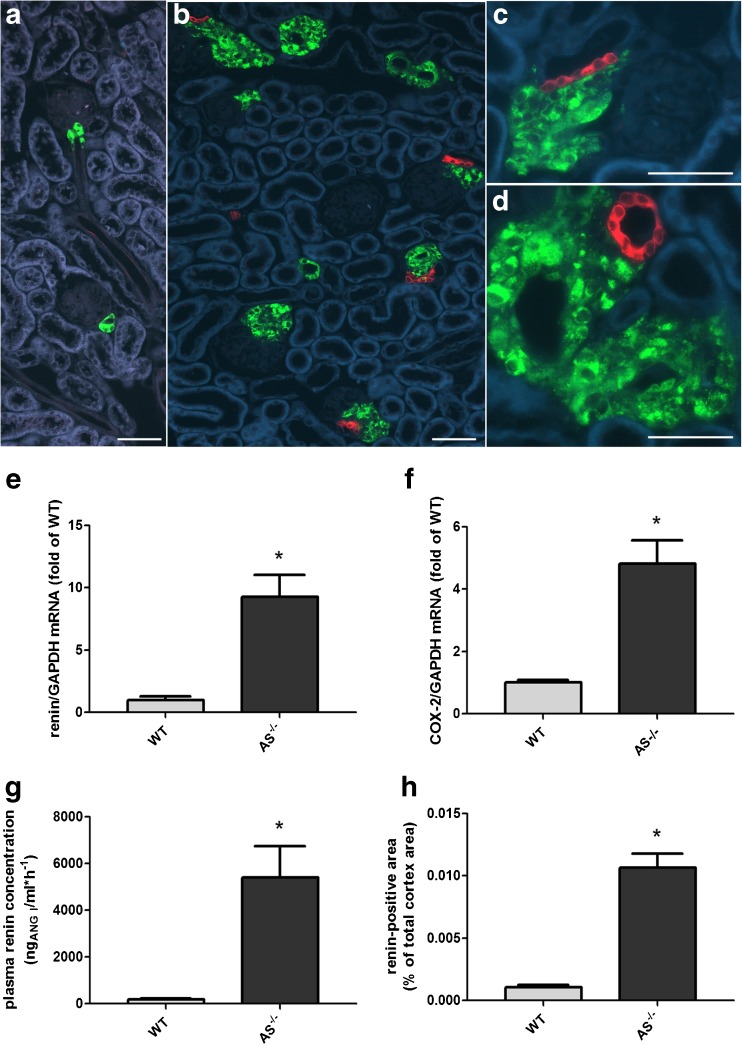
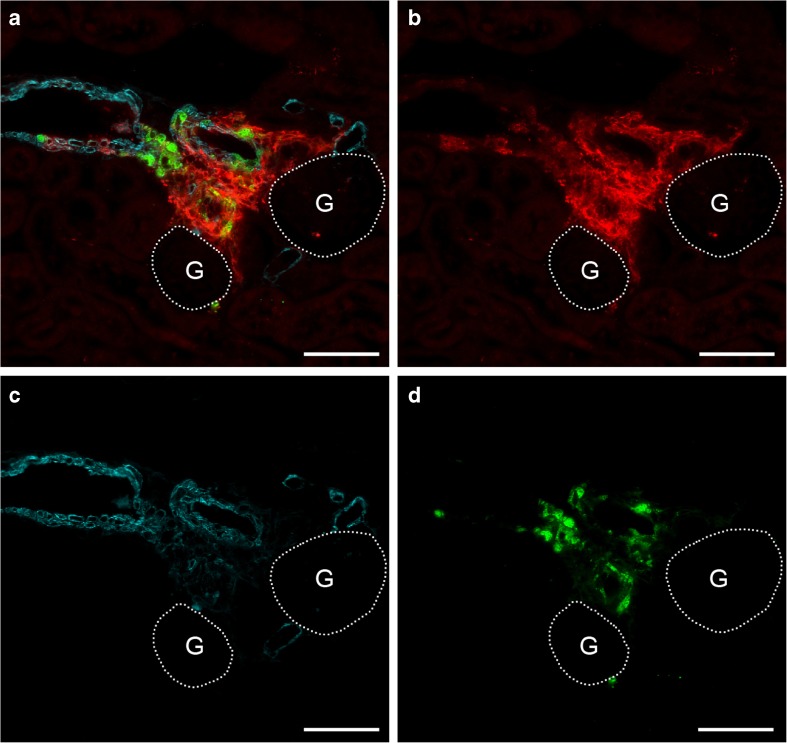
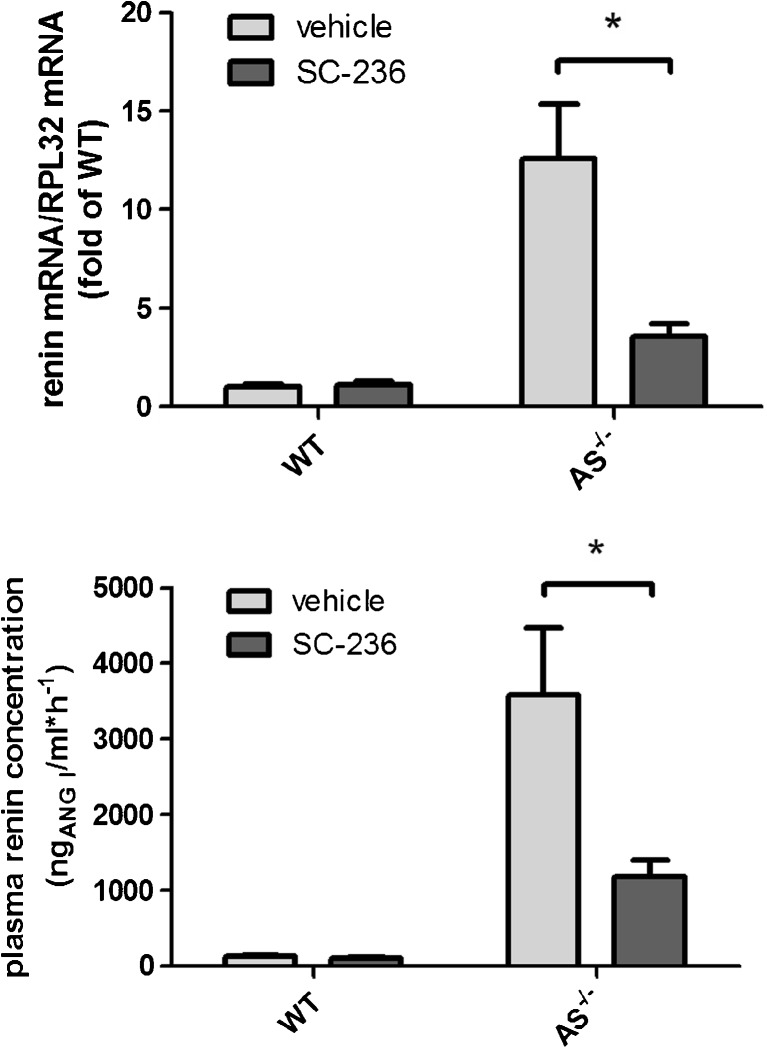
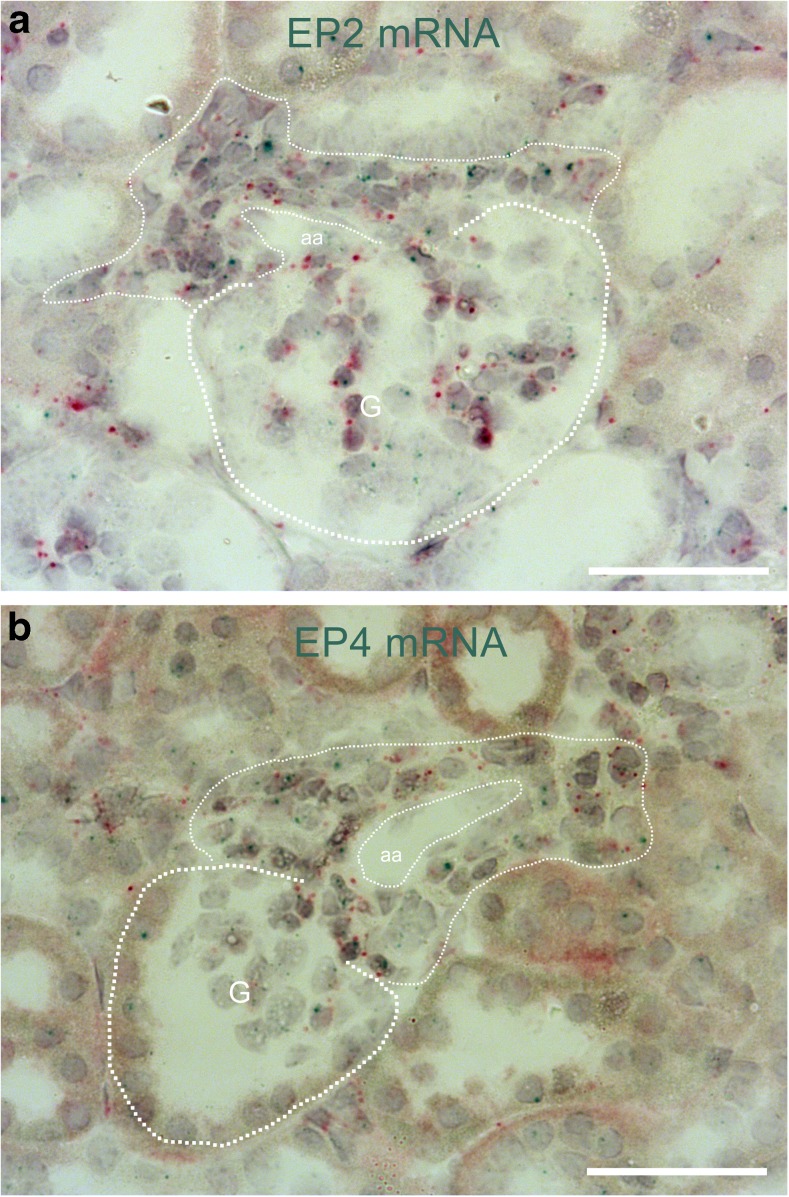
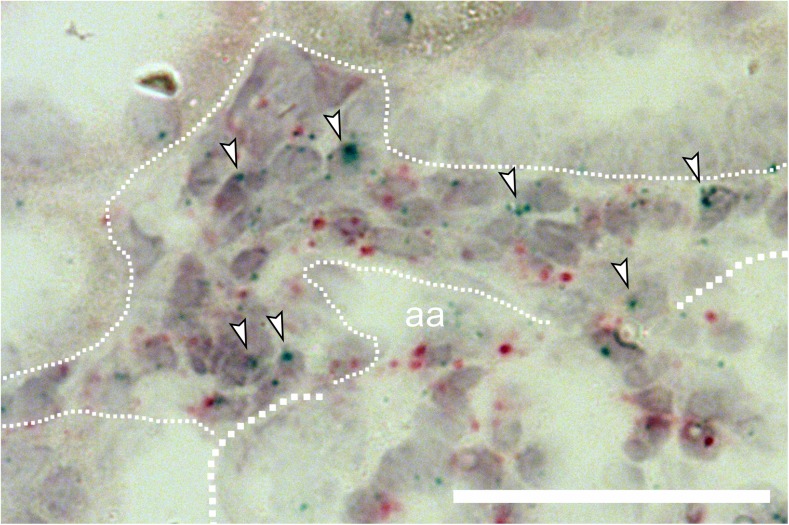
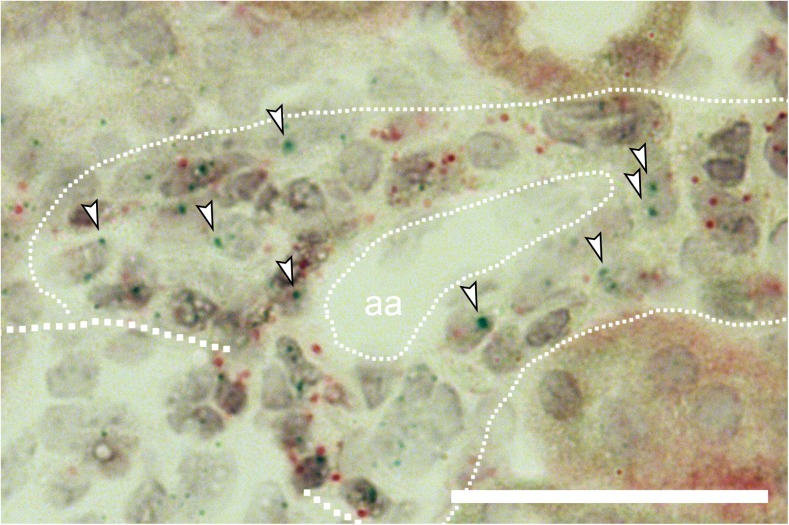
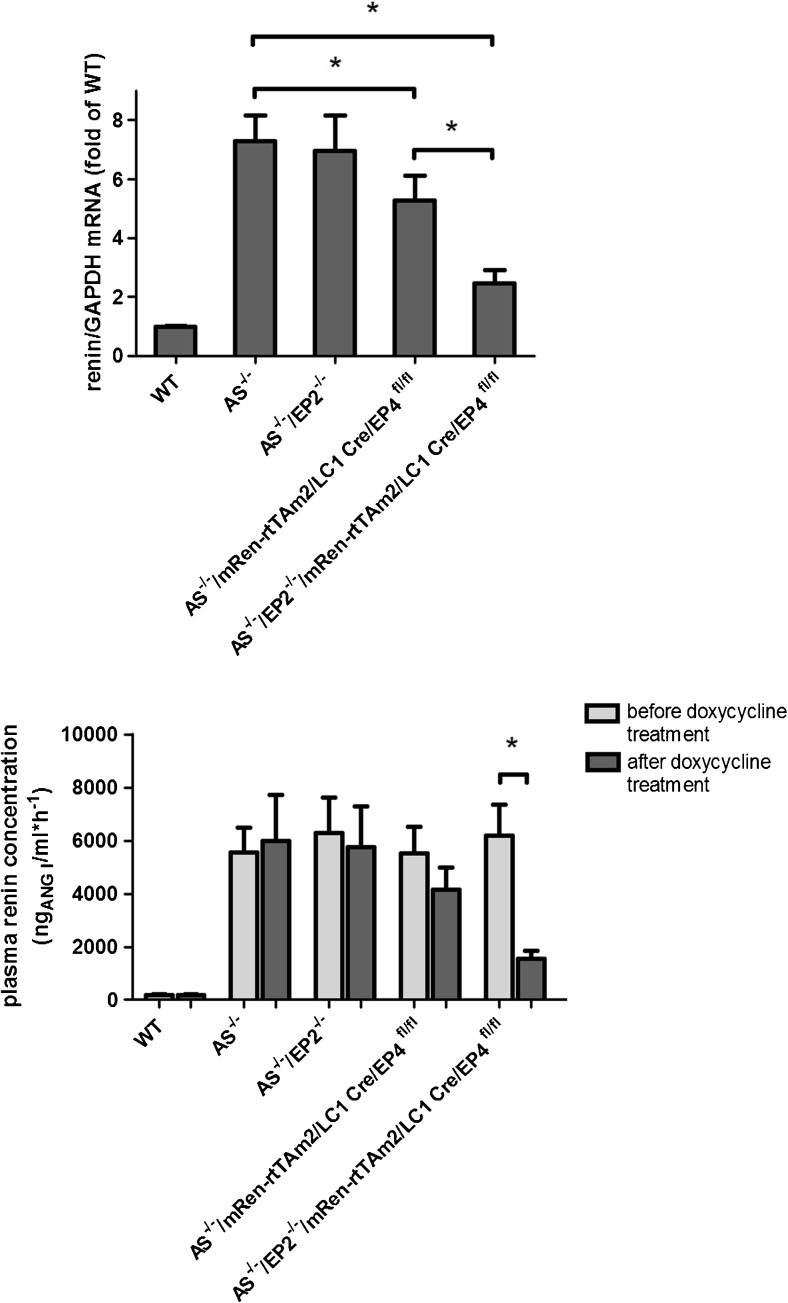
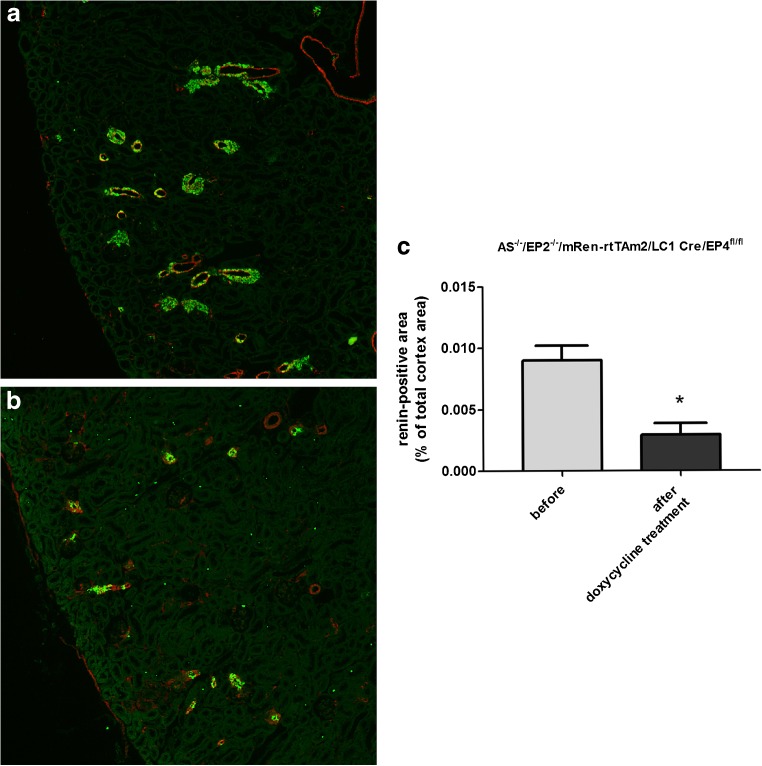
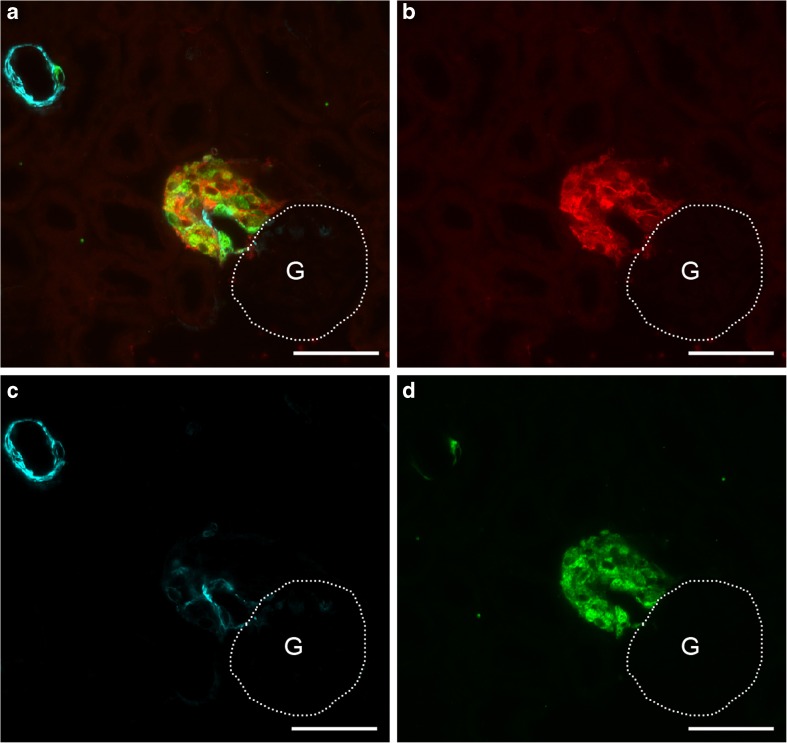
Pharmacological inhibition or genetic loss of function defects of the renin angiotensin aldosterone system (RAAS) causes compensatory renin cell hyperplasia and hyperreninemia. The triggers for the compensatory stimulation of renin synthesis and secretion in this situation may be multimodal. Since cyclooxygenase-2 (COX-2) expression in the macula densa is frequently increased in states of a defective RAAS, we have investigated a potential role of COX-2 and its derived prostaglandins for renin expression and secretion in aldosterone synthase-deficient mice (AS) as a model for a genetic defect of the RAAS. In comparison with wild-type mice (WT), AS mice had 9-fold and 30-fold increases of renin mRNA and of plasma renin concentrations (PRC), respectively. Renin immunoreactivity in the kidney cortex of AS mice was 10-fold higher than in WT. Macula densa COX-2 expression was 5-fold increased in AS kidneys relative to WT kidneys. Treatment of AS mice with the COX-2 inhibitor SC-236 for 1 week lowered both renal renin mRNA and PRC by 70%. Hyperplastic renin cells in AS kidneys were found to express the prostaglandin E receptors EP2 and EP4. Global deletion of EP2 receptors did not alter renin mRNA nor PRC values in AS mice. Renin cell-specific inducible deletion of the EP4 receptor lowered renin mRNA and PRC by 25% in AS mice. Renin cell-specific inducible deletion of the EP4 receptor in combination with global deletion of the EP2 receptor lowered renin mRNA and PRC by 70-75% in AS mice. Lineage tracing of renin-expressing cells revealed that deletion of EP2 and EP4 leads to a preferential downregulation of perivascular renin expression. Our findings suggest that increased macula densa COX-2 activity in AS mice triggers perivascular renin expression and secretion via prostaglandin E.
肾素-血管紧张素-醛固酮系统(RAAS)的药理学抑制或遗传功能缺陷会导致肾素细胞增生和高肾素血症的代偿性。在这种情况下,肾素合成和分泌的代偿性刺激的触发因素可能是多模式的。由于在 RAAS 缺陷状态下,致密斑中的环氧化酶-2(COX-2)表达经常增加,我们研究了 COX-2 及其衍生的前列腺素在醛固酮合酶缺陷型小鼠(AS)中的潜在作用,作为 RAAS 遗传缺陷的模型。与野生型小鼠(WT)相比,AS 小鼠的肾素 mRNA 和血浆肾素浓度(PRC)分别增加了 9 倍和 30 倍。AS 小鼠肾脏皮质中的肾素免疫反应性比 WT 小鼠高 10 倍。AS 肾脏中的致密斑 COX-2 表达比 WT 肾脏高 5 倍。用 COX-2 抑制剂 SC-236 治疗 1 周后,AS 小鼠的肾素 mRNA 和 PRC 分别降低了 70%。AS 肾脏中的增生性肾素细胞表达前列腺素 E 受体 EP2 和 EP4。EP2 受体的全局缺失并未改变 AS 小鼠的肾素 mRNA 或 PRC 值。EP4 受体的肾素细胞特异性诱导缺失使 AS 小鼠的肾素 mRNA 和 PRC 降低了 25%。EP4 受体的肾素细胞特异性诱导缺失与 EP2 受体的全局缺失相结合,使 AS 小鼠的肾素 mRNA 和 PRC 降低了 70-75%。肾素表达细胞的谱系追踪显示,EP2 和 EP4 的缺失导致血管周围肾素表达的优先下调。我们的研究结果表明,AS 小鼠致密斑 COX-2 活性的增加通过前列腺素 E 触发血管周围肾素的表达和分泌。