Sharifi Somayeh, Pakdel Abbas, Ebrahimi Mansour, Reecy James M, Fazeli Farsani Samaneh, Ebrahimie Esmaeil
Department of Animal Science, College of Agriculture, Isfahan University of Technology, Isfahan, Iran.
Department of Animal Science, Iowa State University, Ames, Iowa, United States of America.
PLoS One. 2018 Feb 22;13(2):e0191227. doi: 10.1371/journal.pone.0191227. eCollection 2018.
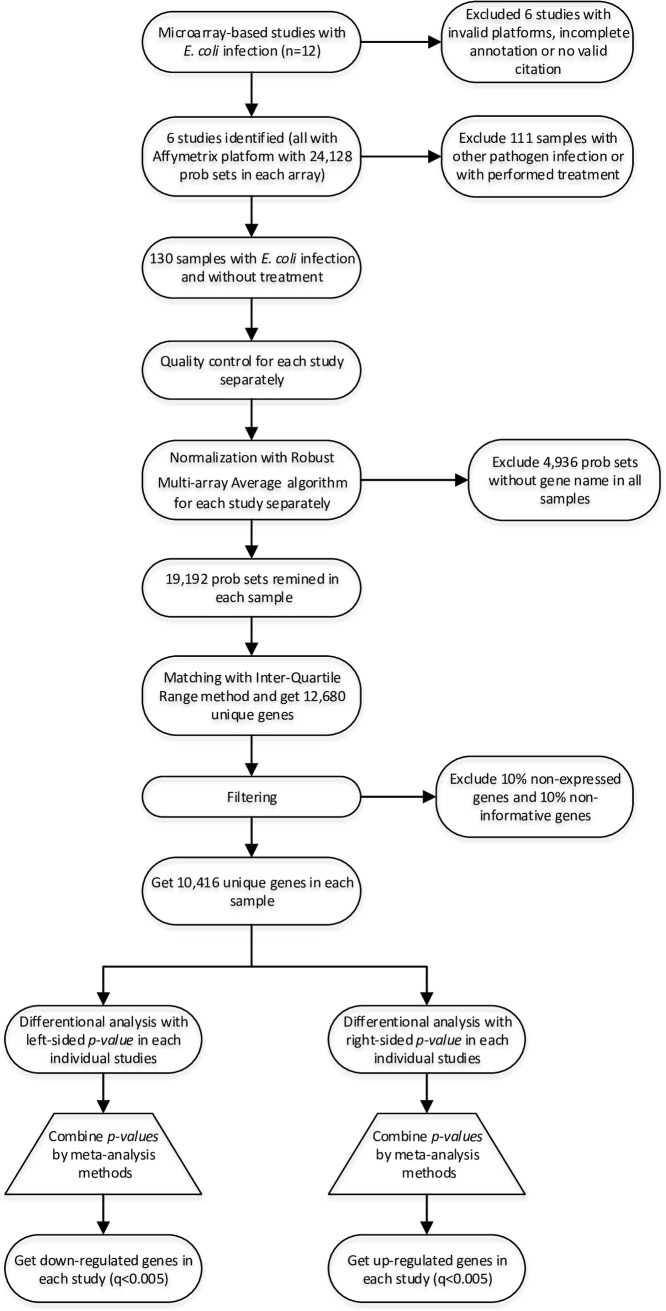
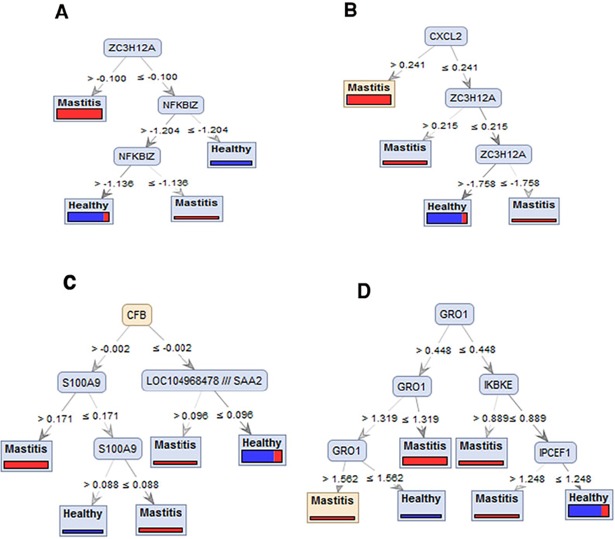
Gram-negative bacteria such as Escherichia coli (E. coli) are assumed to be among the main agents that cause severe mastitis disease with clinical signs in dairy cattle. Rapid detection of this disease is so important in order to prevent transmission to other cows and helps to reduce inappropriate use of antibiotics. With the rapid progress in high-throughput technologies, and accumulation of various kinds of '-omics' data in public repositories, there is an opportunity to retrieve, integrate, and reanalyze these resources to improve the diagnosis and treatment of different diseases and to provide mechanistic insights into host resistance in an efficient way. Meta-analysis is a relatively inexpensive option with good potential to increase the statistical power and generalizability of single-study analysis. In the current meta-analysis research, six microarray-based studies that investigate the transcriptome profile of mammary gland tissue after induced mastitis by E. coli infection were used. This meta-analysis not only reinforced the findings in individual studies, but also several novel terms including responses to hypoxia, response to drug, anti-apoptosis and positive regulation of transcription from RNA polymerase II promoter enriched by up-regulated genes. Finally, in order to identify the small sets of genes that are sufficiently informative in E. coli mastitis, the differentially expressed gene introduced by meta-analysis were prioritized by using ten different attribute weighting algorithms. Twelve meta-genes were detected by the majority of attribute weighting algorithms (with weight above 0.7) as most informative genes including CXCL8 (IL8), NFKBIZ, HP, ZC3H12A, PDE4B, CASP4, CXCL2, CCL20, GRO1(CXCL1), CFB, S100A9, and S100A8. Interestingly, the results have been demonstrated that all of these genes are the key genes in the immune response, inflammation or mastitis. The Decision tree models efficiently discovered the best combination of the meta-genes as bio-signature and confirmed that some of the top-ranked genes -ZC3H12A, CXCL2, GRO, CFB- as biomarkers for E. coli mastitis (with the accuracy 83% in average). This research properly indicated that by combination of two novel data mining tools, meta-analysis and machine learning, increased power to detect most informative genes that can help to improve the diagnosis and treatment strategies for E. coli associated with mastitis in cattle.
革兰氏阴性菌,如大肠杆菌,被认为是导致奶牛出现严重乳腺炎疾病并伴有临床症状的主要病原体之一。快速检测这种疾病对于防止疾病传播给其他奶牛非常重要,并且有助于减少抗生素的不当使用。随着高通量技术的快速发展以及公共数据库中各类“组学”数据的积累,有机会检索、整合和重新分析这些资源,以改进不同疾病的诊断和治疗,并以高效的方式提供宿主抗性的机制性见解。荟萃分析是一种相对廉价的选择,具有提高单研究分析的统计效力和普遍性的良好潜力。在当前的荟萃分析研究中,使用了六项基于微阵列的研究,这些研究调查了大肠杆菌感染诱导乳腺炎后乳腺组织的转录组图谱。这项荟萃分析不仅强化了个别研究的结果,还发现了几个新的术语,包括对缺氧的反应、对药物的反应、抗凋亡以及由上调基因富集的RNA聚合酶II启动子的转录正调控。最后,为了识别在大肠杆菌乳腺炎中具有足够信息的少量基因集,通过使用十种不同的属性加权算法对荟萃分析引入的差异表达基因进行了优先级排序。大多数属性加权算法(权重高于0.7)检测到十二个元基因是最具信息的基因,包括CXCL8(IL8)、NFKBIZ、HP、ZC3H12A、PDE4B、CASP4、CXCL2、CCL20、GRO1(CXCL1)、CFB、S100A9和S100A8。有趣的是,结果表明所有这些基因都是免疫反应、炎症或乳腺炎中的关键基因。决策树模型有效地发现了元基因作为生物标志物的最佳组合,并确认了一些排名靠前的基因——ZC3H12A、CXCL2、GRO、CFB——作为大肠杆菌乳腺炎的生物标志物(平均准确率为83%)。这项研究恰当地表明,通过结合两种新颖的数据挖掘工具——荟萃分析和机器学习,可以提高检测最具信息基因的能力,这有助于改进与奶牛大肠杆菌乳腺炎相关的诊断和治疗策略。