Noel-MacDonnell Janelle R, Usset Joseph, Goode Ellen L, Fridley Brooke L
Department of Biostatistics, University of Kansas Medical Center, Kansas City, KS, United States of America.
Department of Health Services and Outcomes Research, Children's Mercy Hospital, Kansas City, MO, United States of America.
PLoS One. 2018 Feb 27;13(2):e0191758. doi: 10.1371/journal.pone.0191758. eCollection 2018.
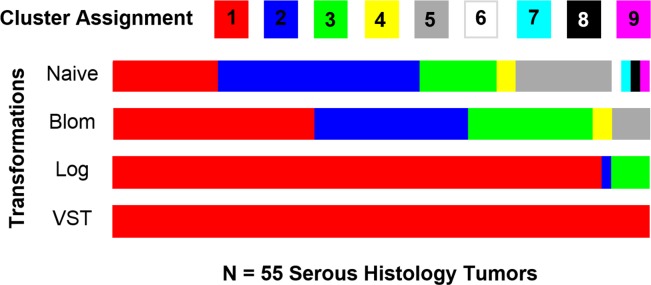
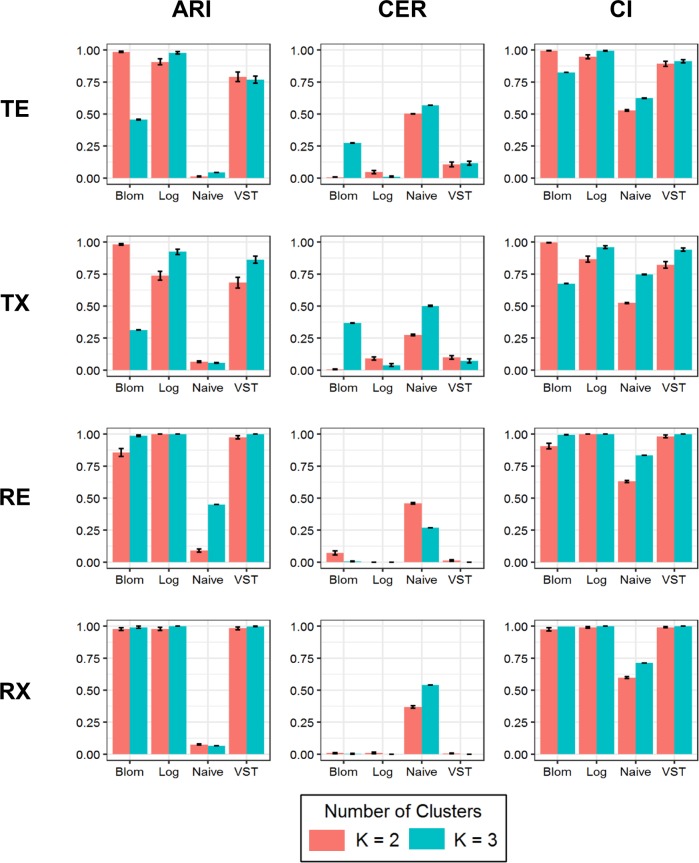
Quality control, global biases, normalization, and analysis methods for RNA-Seq data are quite different than those for microarray-based studies. The assumption of normality is reasonable for microarray based gene expression data; however, RNA-Seq data tend to follow an over-dispersed Poisson or negative binomial distribution. Little research has been done to assess how data transformations impact Gaussian model-based clustering with respect to clustering performance and accuracy in estimating the correct number of clusters in RNA-Seq data. In this article, we investigate Gaussian model-based clustering performance and accuracy in estimating the correct number of clusters by applying four data transformations (i.e., naïve, logarithmic, Blom, and variance stabilizing transformation) to simulated RNA-Seq data. To do so, an extensive simulation study was carried out in which the scenarios varied in terms of: how genes were selected to be included in the clustering analyses, size of the clusters, and number of clusters. Following the application of the different transformations to the simulated data, Gaussian model-based clustering was carried out. To assess clustering performance for each of the data transformations, the adjusted rand index, clustering error rate, and concordance index were utilized. As expected, our results showed that clustering performance was gained in scenarios where data transformations were applied to make the data appear "more" Gaussian in distribution.
RNA测序数据的质量控制、全局偏差、归一化和分析方法与基于微阵列的研究有很大不同。对于基于微阵列的基因表达数据,正态性假设是合理的;然而,RNA测序数据往往遵循过度分散的泊松分布或负二项分布。关于数据转换如何影响基于高斯模型的聚类在RNA测序数据中的聚类性能和估计正确聚类数目的准确性方面,几乎没有研究。在本文中,我们通过对模拟的RNA测序数据应用四种数据转换(即朴素转换、对数转换、布洛姆转换和方差稳定转换)来研究基于高斯模型的聚类在估计正确聚类数目的性能和准确性。为此,我们进行了一项广泛的模拟研究,其中场景在以下方面有所不同:如何选择基因纳入聚类分析、聚类大小和聚类数量。在对模拟数据应用不同的转换之后,进行基于高斯模型的聚类。为了评估每种数据转换的聚类性能,我们使用了调整兰德指数、聚类错误率和一致性指数。正如预期的那样,我们的结果表明,在应用数据转换使数据在分布上显得“更”呈高斯分布的场景中,聚类性能得到了提高。