Center for Human Disease Modeling, Duke University, 300 North Duke Street, Durham, NC, 27701, USA.
Department of Medical Genetics, University of Athens Medical School, Aghia Sophia Children's Hospital, 11527, Athens, Greece.
Hum Genomics. 2018 Mar 1;12(1):11. doi: 10.1186/s40246-018-0141-y.
Intellectual disability (ID) is a common condition with a population prevalence frequency of 1-3% and an enrichment for males, driven in part by the contribution of mutant alleles on the X-chromosome. Among the more than 500 genes associated with ID, DDX3X represents an outlier in sex specificity. Nearly all reported pathogenic variants of DDX3X are de novo, affect mostly females, and appear to be loss of function variants, consistent with the hypothesis that haploinsufficiency at this locus on the X-chromosome is likely to be lethal in males.
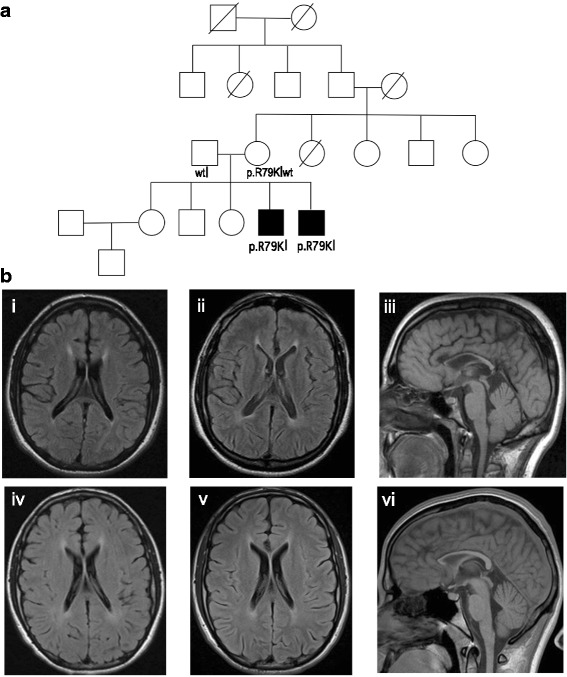
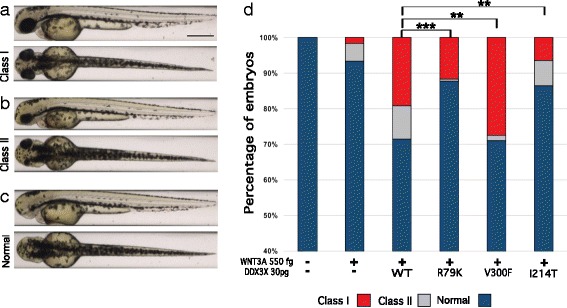
We evaluated two male siblings with syndromic features characterized by mild-to-moderate ID and progressive spasticity. Quad-based whole-exome sequencing revealed a maternally inherited missense variant encoding p.R79K in DDX3X in both siblings and no other apparent pathogenic variants. We assessed its possible relevance to their phenotype using an established functional assay for DDX3X activity in zebrafish embryos and found that this allele causes a partial loss of DDX3X function and thus represents a hypomorphic variant.
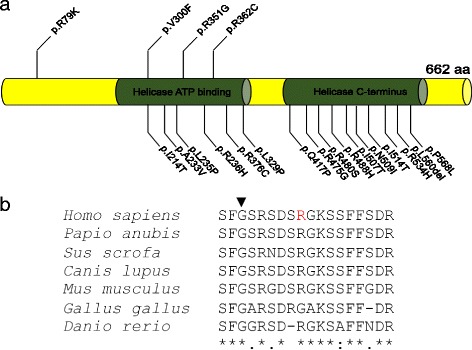
Our genetic and functional data suggest that partial loss of function of DDX3X can cause syndromic ID. The p.R79K allele affects a region of the protein outside the critical RNA helicase domain, offering a credible explanation for the observed retention of partial function, viability in hemizygous males, and lack of pathology in females. These findings expand the gender spectrum of pathology of this locus and suggest that analysis for DDX3X variants should be considered relevant for both males and females.
智力障碍(ID)是一种常见病症,其人群患病率为 1-3%,且男性发病率更高,部分原因是 X 染色体上突变等位基因的贡献。在与 ID 相关的 500 多个基因中,DDX3X 是性别特异性的一个例外。几乎所有报道的 DDX3X 致病变体都是新生的,主要影响女性,且似乎是功能丧失变体,这与 X 染色体上该基因座的杂合不足在男性中可能是致命的假设一致。
我们评估了两名具有综合征特征的男性兄弟,其特征为轻度至中度 ID 和进行性痉挛。基于四联体的全外显子组测序在这对兄弟中均发现了一个母系遗传的错义变体,该变体在 DDX3X 中编码 p.R79K,且没有其他明显的致病性变体。我们使用已建立的斑马鱼胚胎中 DDX3X 活性的功能测定来评估其对他们表型的可能相关性,发现该等位基因导致 DDX3X 功能部分丧失,因此代表了一个功能减弱的变体。
我们的遗传和功能数据表明,DDX3X 的部分功能丧失可导致综合征性 ID。p.R79K 等位基因影响蛋白的关键 RNA 解旋酶结构域外的区域,为观察到的部分功能保留、半合子男性的存活和女性无病理学提供了可信的解释。这些发现扩展了该基因座病理学的性别范围,并表明应考虑对男性和女性进行 DDX3X 变体分析。