Smirnov Alexey, Zubrienė Asta, Manakova Elena, Gražulis Saulius, Matulis Daumantas
Department of Biothermodynamics and Drug Design, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania.
Department of Protein-DNA Interactions, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania.
PeerJ. 2018 Feb 26;6:e4412. doi: 10.7717/peerj.4412. eCollection 2018.
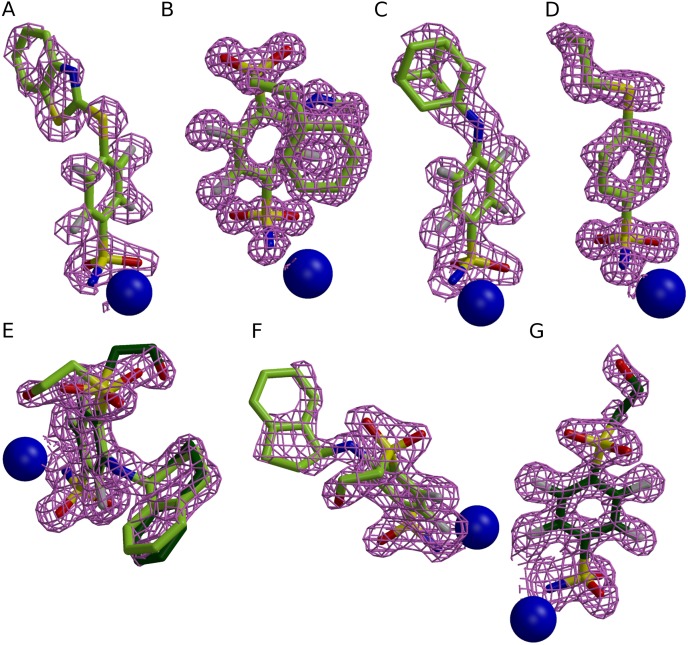
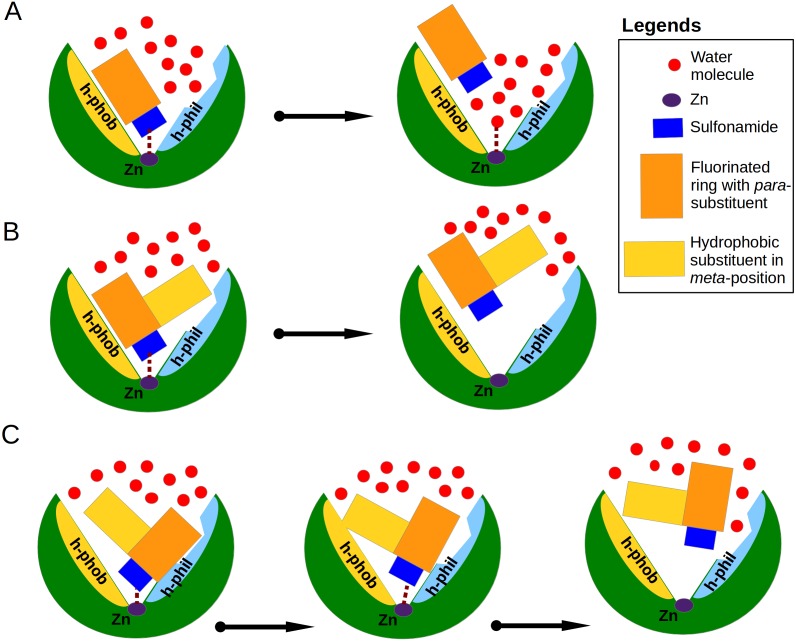
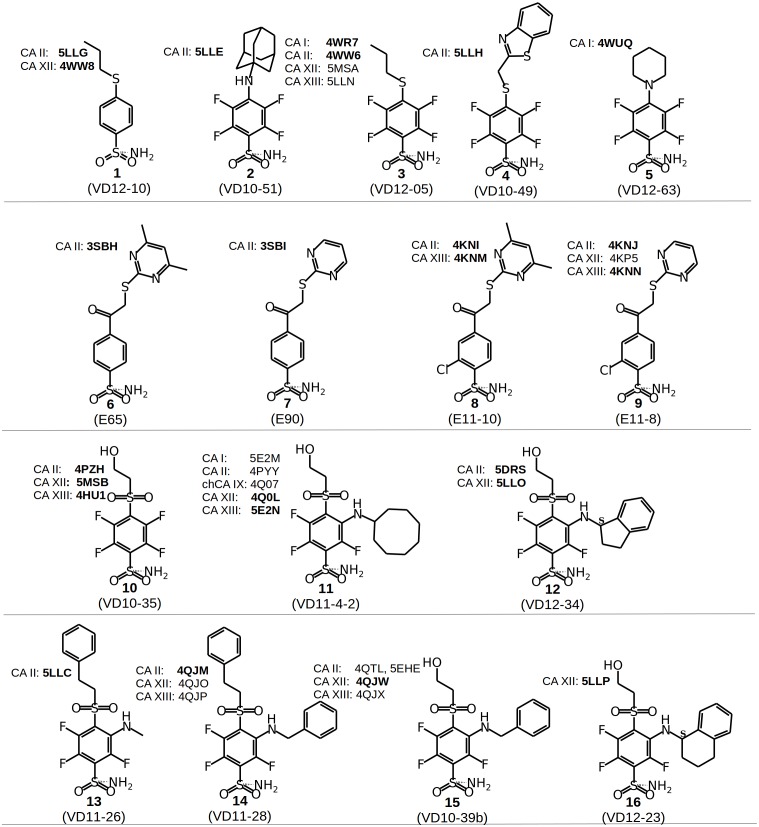
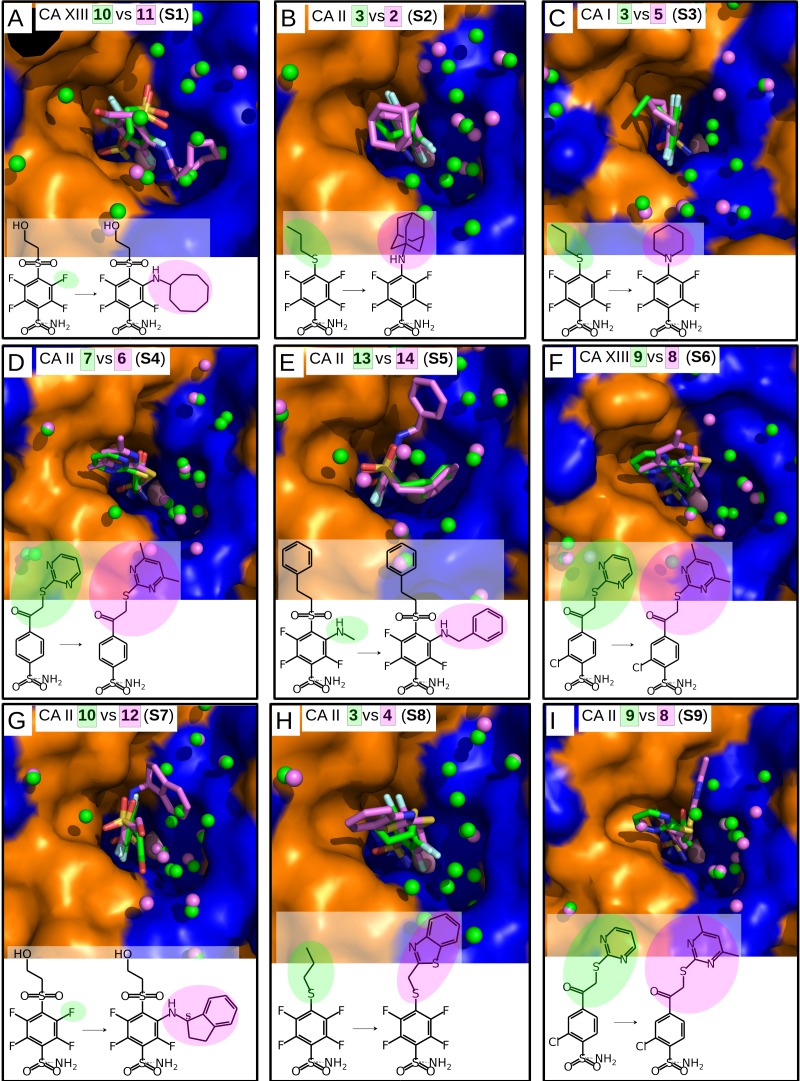
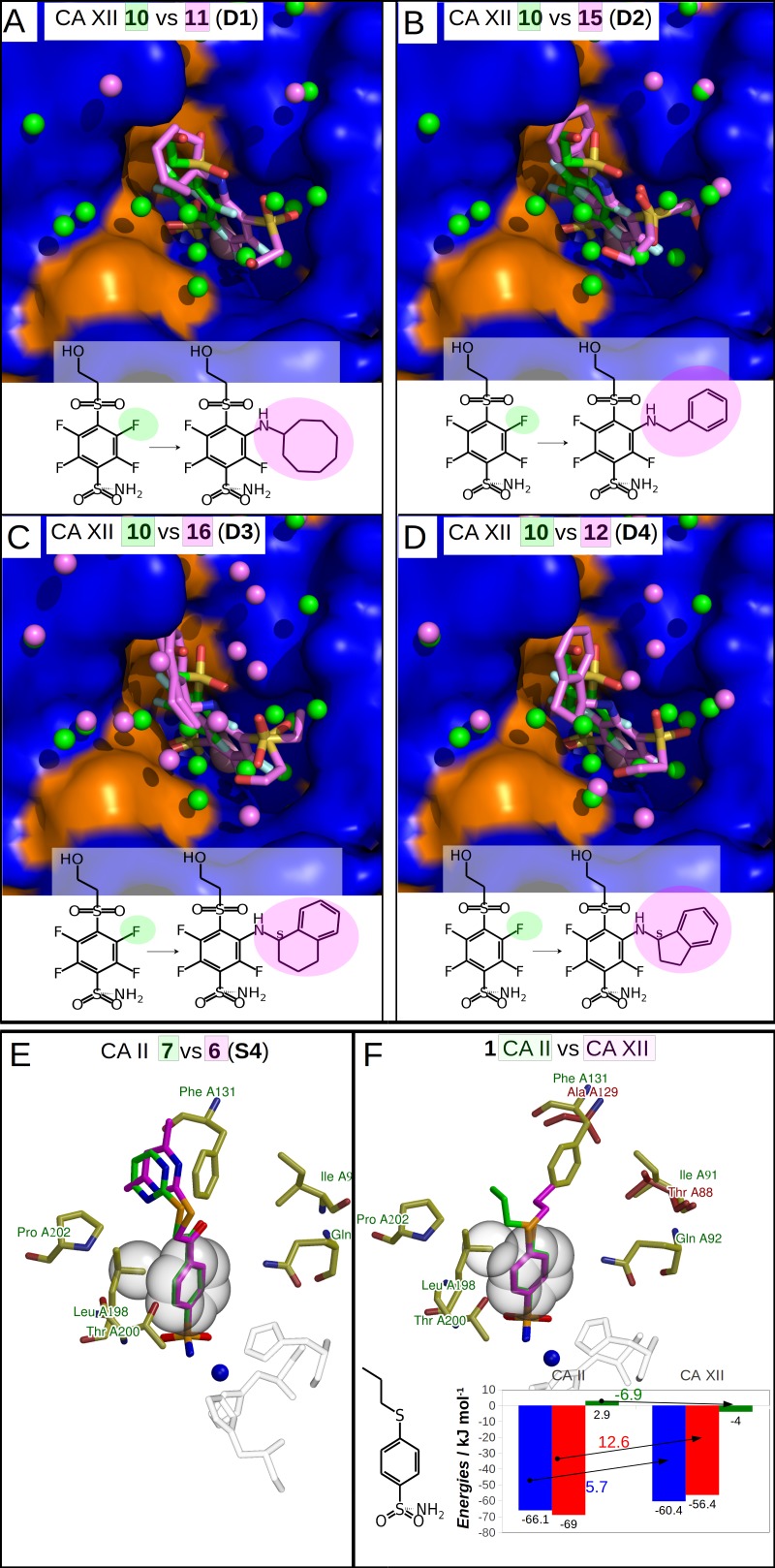
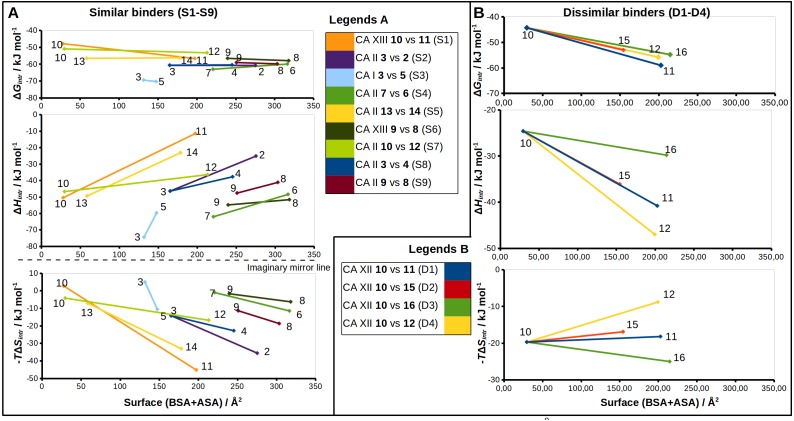
The structure-thermodynamics correlation analysis was performed for a series of fluorine- and chlorine-substituted benzenesulfonamide inhibitors binding to several human carbonic anhydrase (CA) isoforms. The total of 24 crystal structures of 16 inhibitors bound to isoforms CA I, CA II, CA XII, and CA XIII provided the structural information of selective recognition between a compound and CA isoform. The binding thermodynamics of all structures was determined by the analysis of binding-linked protonation events, yielding the intrinsic parameters, i.e., the enthalpy, entropy, and Gibbs energy of binding. Inhibitor binding was compared within structurally similar pairs that differ by or -substituents enabling to obtain the contributing energies of ligand fragments. The pairs were divided into two groups. First, binders-the pairs that keep the same orientation of the benzene ring exhibited classical hydrophobic effect, a less exothermic enthalpy and a more favorable entropy upon addition of the hydrophobic fragments. Second, binders-the pairs of binders that demonstrated altered positions of the benzene rings exhibited the non-classical hydrophobic effect, a more favorable enthalpy and variable entropy contribution. A deeper understanding of the energies contributing to the protein-ligand recognition should lead toward the eventual goal of rational drug design where chemical structures of ligands could be designed based on the target protein structure.
对一系列与几种人类碳酸酐酶(CA)同工型结合的氟代和氯代苯磺酰胺抑制剂进行了结构 - 热力学相关性分析。16种抑制剂与CA I、CA II、CA XII和CA XIII同工型结合的总共24个晶体结构提供了化合物与CA同工型之间选择性识别的结构信息。通过分析结合相关的质子化事件确定了所有结构的结合热力学,得出了内在参数,即结合焓、熵和吉布斯自由能。在由 或 -取代基不同的结构相似对中比较抑制剂结合情况,从而获得配体片段的贡献能量。这些对被分为两组。第一组, 结合剂——苯环保持相同取向的对表现出经典疏水效应,添加疏水片段时焓变放热较少且熵变更有利。第二组, 结合剂——苯环位置发生改变的结合剂对表现出非经典疏水效应,焓变更有利且熵变贡献可变。对有助于蛋白质 - 配体识别的能量有更深入的理解应该朝着合理药物设计的最终目标迈进,即在基于靶蛋白结构设计配体化学结构。