The Jackson Laboratory for Mammalian Genetics, Bar Harbor, Maine 04609.
The Jackson Laboratory for Genomic Medicine, Farmington, Connecticut 06032.
Genetics. 2018 May;209(1):51-64. doi: 10.1534/genetics.117.300673. Epub 2018 Mar 5.
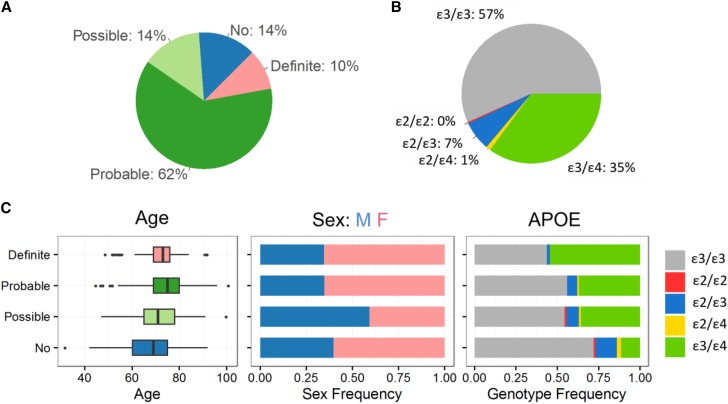
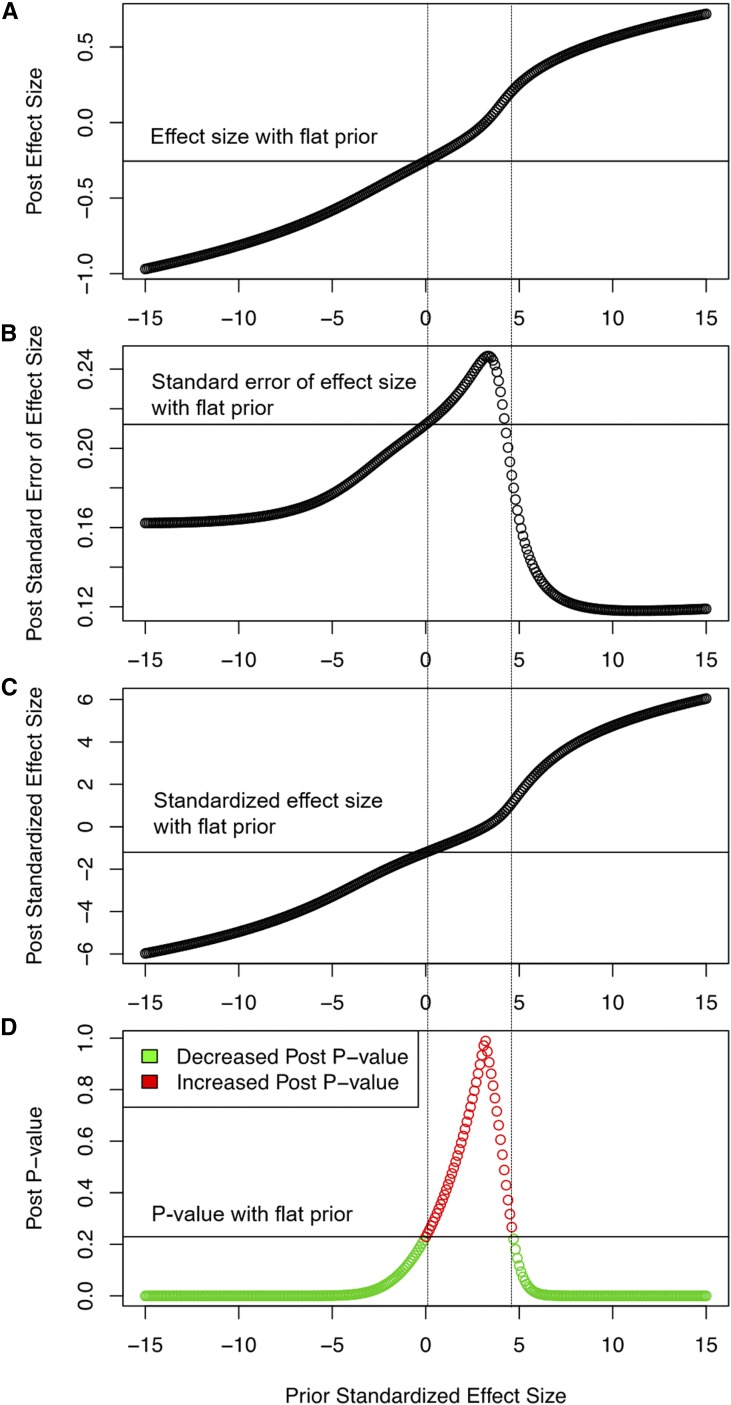
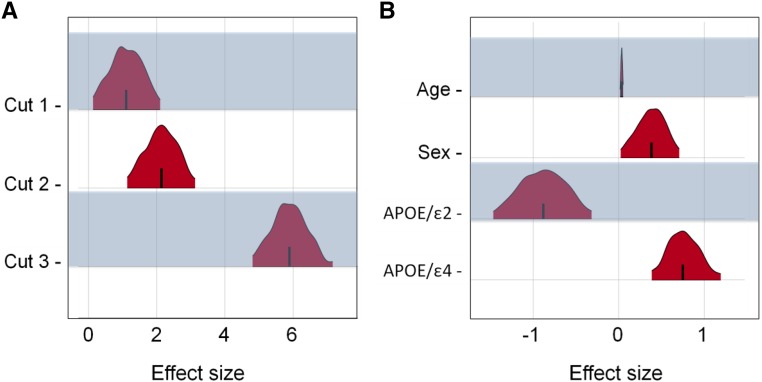
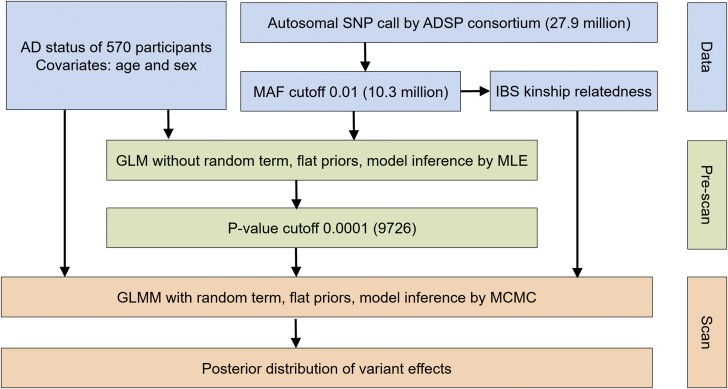
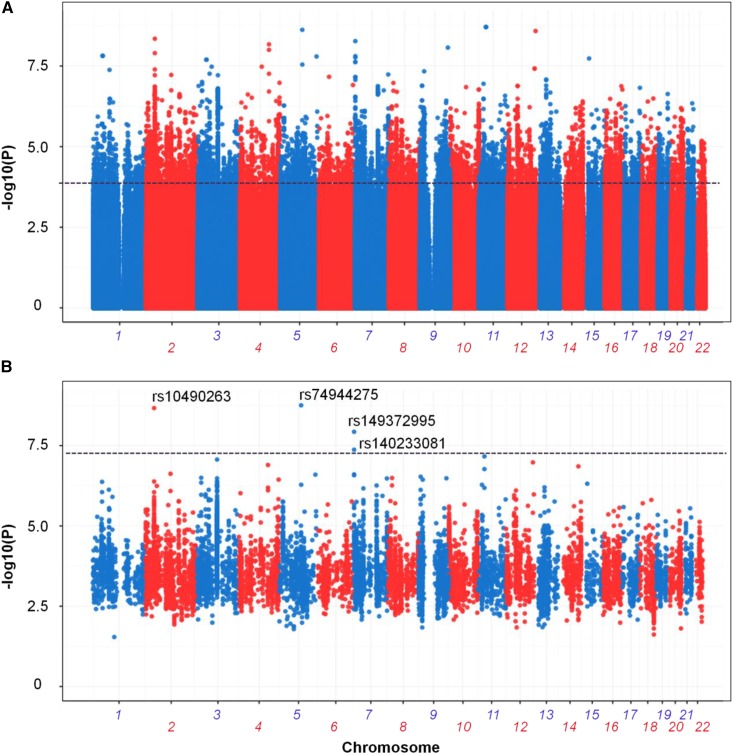
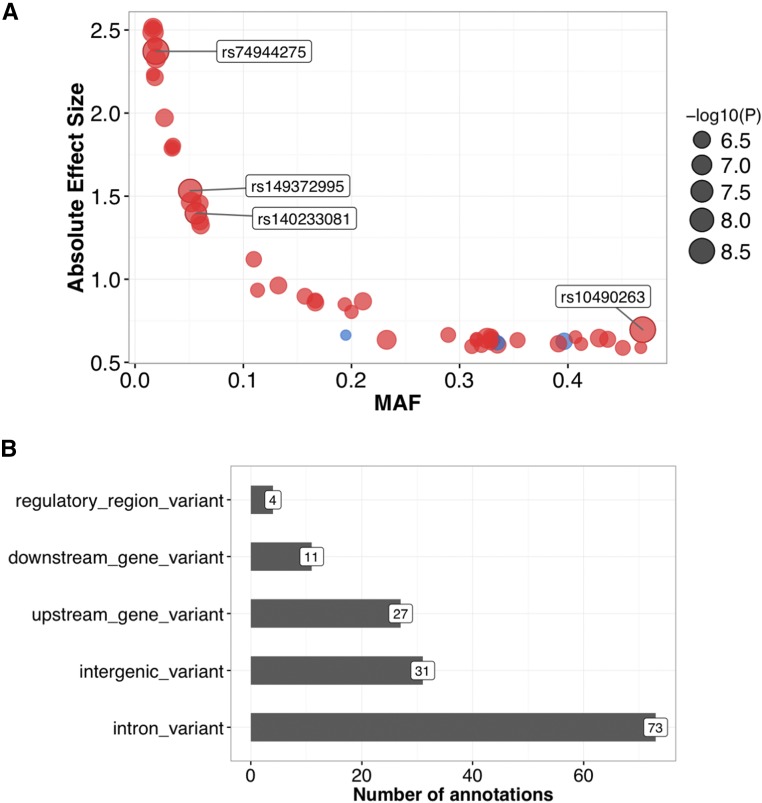
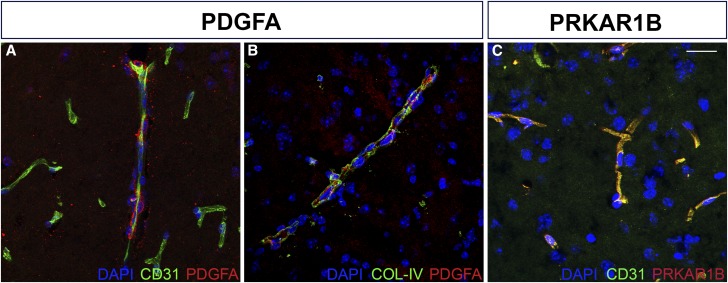
Recent technical and methodological advances have greatly enhanced genome-wide association studies (GWAS). The advent of low-cost, whole-genome sequencing facilitates high-resolution variant identification, and the development of linear mixed models (LMM) allows improved identification of putatively causal variants. While essential for correcting false positive associations due to sample relatedness and population stratification, LMMs have commonly been restricted to quantitative variables. However, phenotypic traits in association studies are often categorical, coded as binary case-control or ordered variables describing disease stages. To address these issues, we have devised a method for genomic association studies that implements a generalized LMM (GLMM) in a Bayesian framework, called Bayes-GLMM has four major features: (1) support of categorical, binary, and quantitative variables; (2) cohesive integration of previous GWAS results for related traits; (3) correction for sample relatedness by mixed modeling; and (4) model estimation by both Markov chain Monte Carlo sampling and maximal likelihood estimation. We applied Bayes-GLMM to the whole-genome sequencing cohort of the Alzheimer's Disease Sequencing Project. This study contains 570 individuals from 111 families, each with Alzheimer's disease diagnosed at one of four confidence levels. Using Bayes-GLMM we identified four variants in three loci significantly associated with Alzheimer's disease. Two variants, rs140233081 and rs149372995, lie between and The coded proteins are localized to the glial-vascular unit, and transcript levels are associated with Alzheimer's disease-related neuropathology. In summary, this work provides implementation of a flexible, generalized mixed-model approach in a Bayesian framework for association studies.
最近的技术和方法上的进步极大地增强了全基因组关联研究(GWAS)。低成本、全基因组测序的出现促进了高分辨率变异的识别,而线性混合模型(LMM)的发展则允许更有效地识别潜在的因果变异。虽然对于纠正由于样本相关性和群体分层引起的假阳性关联至关重要,但 LMM 通常仅限于定量变量。然而,关联研究中的表型特征通常是分类的,编码为二进制病例对照或描述疾病阶段的有序变量。为了解决这些问题,我们设计了一种用于基因组关联研究的方法,该方法在贝叶斯框架中实现了广义线性混合模型(GLMM),称为 Bayes-GLMM。Bayes-GLMM 有四个主要特点:(1)支持分类、二进制和定量变量;(2)对相关性状的先前 GWAS 结果进行整合;(3)通过混合建模纠正样本相关性;(4)通过马尔可夫链蒙特卡罗抽样和最大似然估计进行模型估计。我们将 Bayes-GLMM 应用于阿尔茨海默病测序项目的全基因组测序队列。这项研究包含了 570 名来自 111 个家庭的个体,每个家庭的个体都被诊断为四种置信水平之一的阿尔茨海默病。使用 Bayes-GLMM,我们在三个基因座中鉴定出四个与阿尔茨海默病显著相关的变体。两个变体 rs140233081 和 rs149372995 位于 和 之间。编码的蛋白质定位于神经胶质-血管单元,而 转录水平与阿尔茨海默病相关的神经病理学相关。总之,这项工作为关联研究提供了一种灵活的、广义的混合模型方法在贝叶斯框架中的实现。