Jiangsu Key Laboratory of Neurogeneration, Department of Pharmacology, Nanjing Medical University, 101 Nongmian Avenue, Nanjing, 211166, P.R. China.
Department of Pharmacology, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing, 210023, P.R. China.
Cell Death Dis. 2018 Mar 14;9(3):404. doi: 10.1038/s41419-018-0437-9.
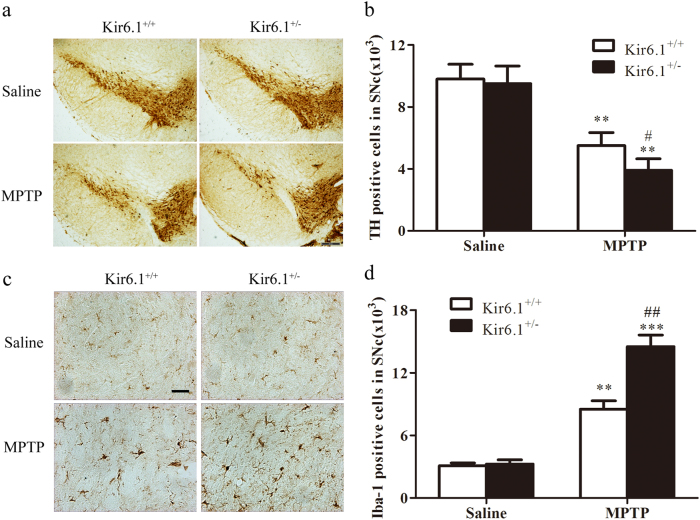
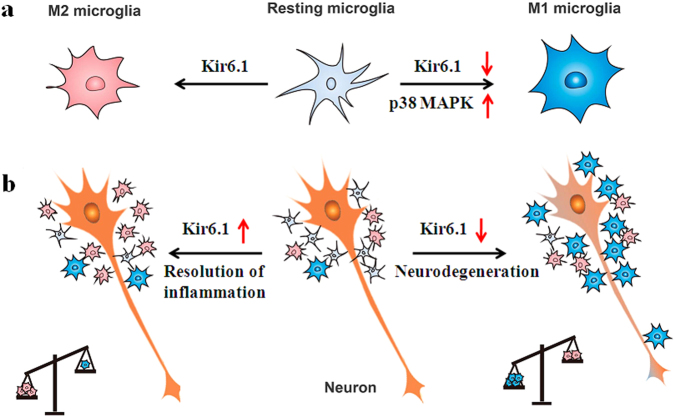
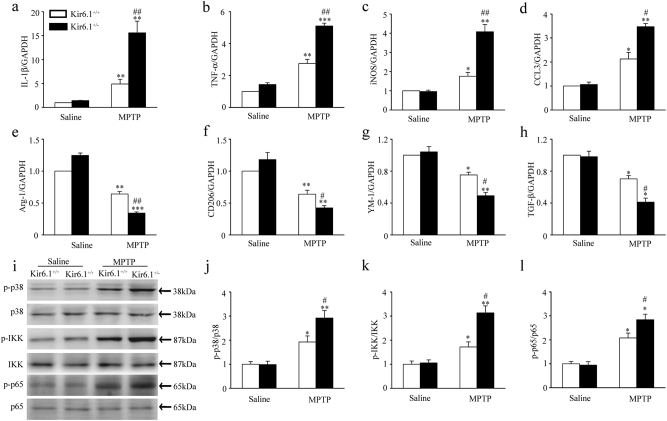
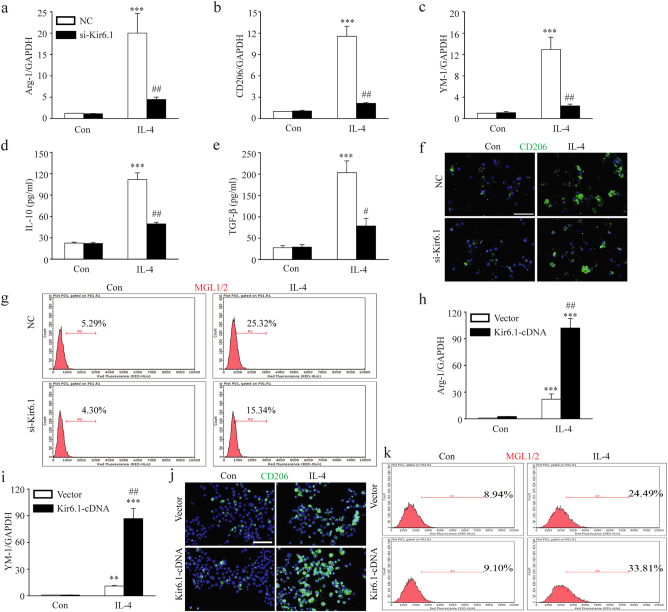
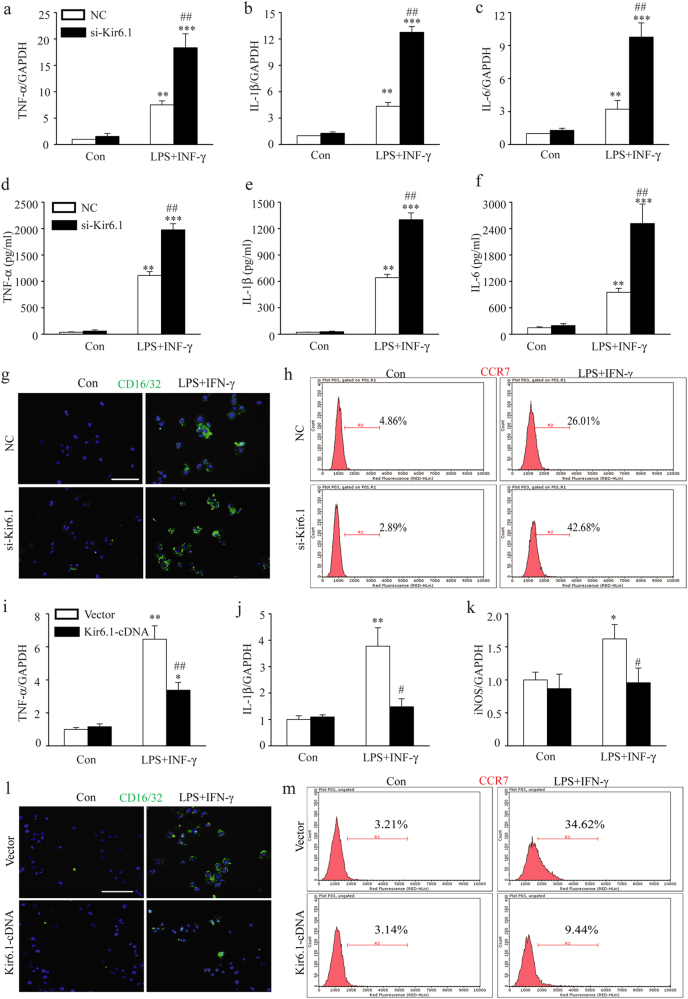
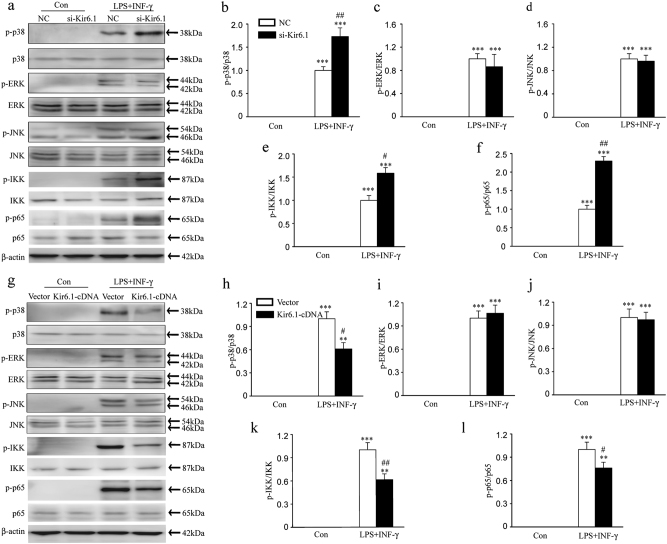
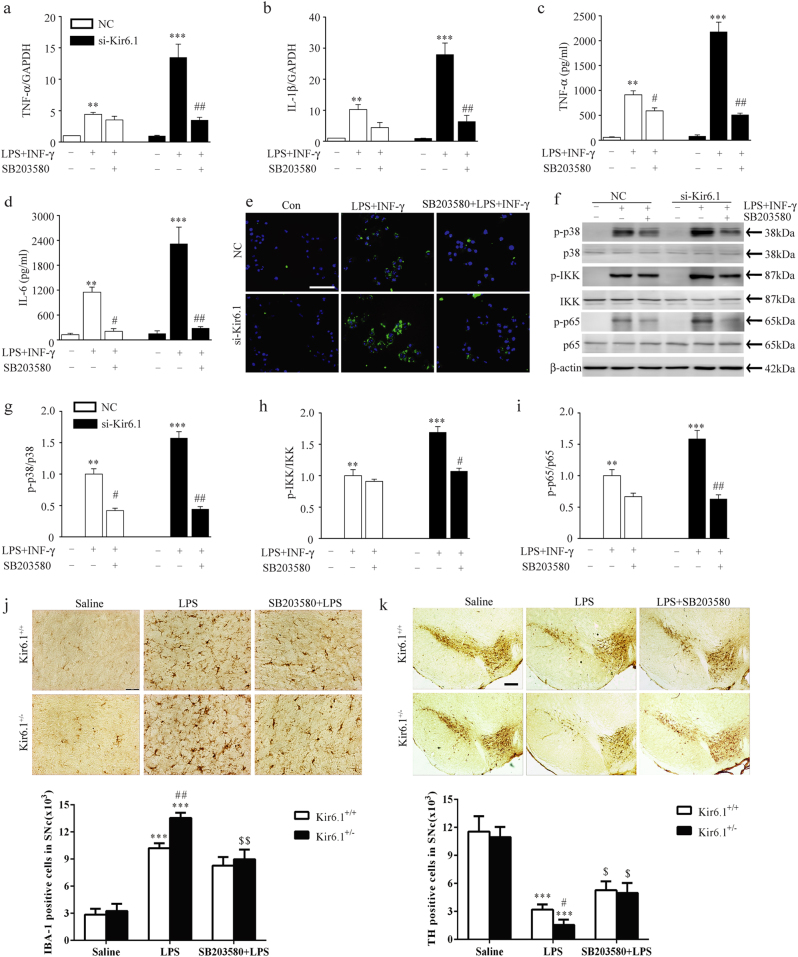
Classical activation (M1 phenotype) and alternative activation (M2 phenotype) are the two polars of microglial activation states that can produce either neurotoxic or neuroprotective effects in the immune pathogenesis of Parkinson's disease (PD). Exploiting the beneficial properties of microglia cells by modulating their polarization states provides great potential for the treatment of PD. However, the mechanism that regulates microglia polarization remains elusive. Here we demonstrated that Kir6.1-containing ATP-sensitive potassium (Kir6.1/K-ATP) channel switched microglia from the detrimental M1 phenotype toward the beneficial M2 phenotype. Kir6.1 knockdown inhibited M2 polarization and simultaneously exaggerated M1 microglial inflammatory responses, while Kir6.1 overexpression promoted M2 polarization and synchronously alleviated the toxic phase of M1 microglia polarization. Furthermore, we observed that the Kir6.1 deficiency dramatically exacerbated dopaminergic neuron death companied by microglia activation in mouse model of PD. Mechanistically, Kir6.1 deficiency enhanced the activation of p38 MAPK-NF-κB pathway and increased the ratio of M1/M2 markers in the substantia nigra compacta of mouse model of PD. Suppression of p38 MAPK in vivo partially rescued the deleterious effects of Kir6.1 ablation on microglia phenotype and dopaminergic neuron death. Collectively, our findings reveal that Kir6.1/K-ATP channel modulates microglia phenotypes transition via inhibition of p38 MAPK-NF-κB signaling pathway and Kir6.1/K-ATP channel may be a promising therapeutic target for PD.
经典激活(M1 表型)和替代激活(M2 表型)是小胶质细胞激活状态的两个极端,它们在帕金森病(PD)的免疫发病机制中既可以产生神经毒性作用,也可以产生神经保护作用。通过调节小胶质细胞的极化状态来利用小胶质细胞的有益特性,为 PD 的治疗提供了巨大的潜力。然而,调节小胶质细胞极化的机制仍不清楚。在这里,我们证明了含有 Kir6.1 的三磷酸腺苷敏感性钾(Kir6.1/K-ATP)通道使小胶质细胞从有害的 M1 表型向有益的 M2 表型转变。Kir6.1 敲低抑制 M2 极化,同时加剧 M1 小胶质细胞炎症反应,而 Kir6.1 过表达促进 M2 极化,同时减轻 M1 小胶质细胞极化的毒性阶段。此外,我们观察到 Kir6.1 缺陷在 PD 小鼠模型中显著加剧了多巴胺能神经元死亡伴小胶质细胞激活。在机制上,Kir6.1 缺陷增强了 p38 MAPK-NF-κB 通路的激活,并增加了 PD 小鼠模型中黑质致密部 M1/M2 标志物的比值。体内抑制 p38 MAPK 部分挽救了 Kir6.1 消融对小胶质细胞表型和多巴胺能神经元死亡的有害影响。总之,我们的研究结果表明,Kir6.1/K-ATP 通道通过抑制 p38 MAPK-NF-κB 信号通路调节小胶质细胞表型的转变,Kir6.1/K-ATP 通道可能是 PD 的一个有前途的治疗靶点。